Система свертывания крови
Механизм гемокоагуляции
Основы ферментной теории свертывания крови были заложены еще в XIX в. профессором Юрьевского университета А. А. Шмидтом (1861 г.; 1895 г.) и уточнены П. Моравитцем в 1905 г. Согласно данной теории образование волокон фибрина, составляющих каркас любого свертка крови, связано с ферментным отщеплением от молекул фибриногена небольших фрагментов (фибринопептидов), после чего остающиеся основные части этих молекул (фибрин‑мономеры) соединяются друг с другом в длинные цепи «фибринполимера».
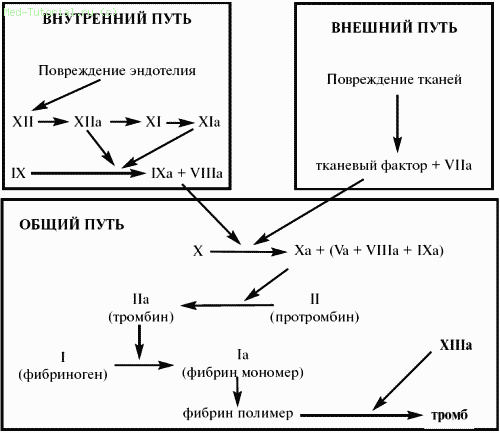
Фермент крови, обеспечивающий отщепление фибрино‑пептидов и превращение фибриногена в фибрин, получил название тромбина. Готового тромбина в плазме нет, но в ней имеется его неактивный предшественник – протромбин (фактор II), который в присутствии ионов кальция и под влиянием «тромбокиназы» превращается в тромбин.
Имеется 2 различных механизма активации свертывания крови. Один из них обозначается как «внешний механизм», поскольку запускается поступлением из тканей или из лейкоцитов в плазму тканевого тромбопластического фактора (фактора III), относящегося к липопротеидам. Этот фактор вступает во взаимодействие с фактором VII и при участии ионов кальция быстро образует активатор фактора X, который и является главной составной частью протромбиназы, поскольку трансформирует протромбин (фактор II) в тромбин (IIа). В лабораторных условиях этот путь имитируется протромбиновым тестом Квика: к исследуемой рекальцифицированной плазме добавляется стандартная доза тканевого (мозгового) тромбо‑пластина, получается, что процесс искусственно запускается по внешнему механизму.
Второй путь активации свертывания назван внутренним, поскольку осуществляется без добавления извне тканевого тромбопластина, за счет внутренних ресурсов плазмы. В искусственных условиях свертывание по внутреннему механизму наблюдается тогда, когда кровь, извлеченная из сосудистого русла, самопроизвольно свертывается в пробирке. Запуск этого внутреннего механизма начинается с активации фактора XII (фактора Хагемана). Эта активация возникает в разных условиях: вследствие контакта крови с поврежденной сосудистой стенкой (коллагеном и другими структурами), с измененными клеточными мембранами, под влиянием некоторых протеаз и адреналина, а вне организма – вследствие контакта крови или плазмы с чужеродной поверхностью – стеклом, иглами, кюветами и др. Этой контактной активации не препятствует удаление из крови ионов кальция, в связи с чем она происходит и в цитратной (или оксалатной) плазме. Однако в этом случае процесс обрывается на активации фактора IX, для которой уже необходим ионизированный кальций. Вслед за фактором XII последовательно активируются факторы XI, IX и VIII. Последние два фактора образуют продукт, который активирует фактор X, что приводит к формированию протромбиназной активности. Вместе с тем сам по себе активированный фактор X обладает слабой протромбиназной активностью, но она усиливается в 1000 раз акселерирующим фактором – фактором V.
Точно так же действие фактора IX на фактор X усиливается в несколько тысяч раз фактором VIII – антигемофильным глобулином. Этим обосновывается деление плазменных факторов свертывания на 2 группы: ферментную – факторы XII, XI, IX, VII, X и II и неферментную – факторы I, V и VIII. Фактор X последовательно отщепляет от протромбина два фрагмента, в результате чего образуется тромбин‑эстераза, отщепляющая от α– и β‑цепей фибриногена вначале 2 пептида А, затем – 2 пептида В (всего 4 фибринопептида). Незавершенный фибрин‑мономер, от которого отделились лишь пептиды А, обозначается как «дес‑А‑фибрин», а лишенный пептидов А и В – как «дес‑АВ‑фибрин». Фибрин‑мономеры имеют трехмодулярную структуру, их сборка в полимер проходит этапы формирования димеров, из которых путем дальнейшего продольного и поперечного связывания образуются протофибрилы фибрина. Соединяясь друг с другом, протофибрилы формируют волокна фибрина. Фибринстабилизирующий фактор XIII (плазменная трансглутаминаза) «прошивает» фибрин‑полимеры дополнительными перекрестными связями между γ‑цепями и тем самым укрепляет фибрин, делает его нерастворимым в мочевине, монохлоруксусной кислоте и других растворителях. Основным активатором фактора XIII является тромбин.
В условиях патологии процесс полимеризации фибрина легко нарушается либо вследствие плохой трансформации дес‑А‑фибрина в дес‑АВ‑фибрин, либо из‑за нарушения сборки димеров и протофибрил. В этих случаях фибрин‑мономеры (дес‑А‑фибрин и дес‑АВ‑фибрин) соединяются с фибриногеном, образуя средне– и крупномолекулярные (от 450 000 до 2 000 000 и более) растворимые фибрин‑мономерные комплексы. Фибриноген в этих комплексах блокируется и утрачивает способность свертывания под влиянием тромбина. Этот феномен, имеющий большое диагностическое значение, в литературе обозначается по‑разному – «растворимые фибрин‑мономерные комплексы» (РФМК), «фибринемия», «несвертывающийся фибрин», «заблокированный, или тромбинрезистентный, фибриноген», «феномен паракоагуляции». Последнее название связано с тем, что не свертывающиеся тромбином РФМК коагулируют или преципитируют под влиянием ряда неферментных воздействий – при добавлении к плазме спирта (этаноловый тест), сульфата протамина (протамин‑сульфатный тест) или при охлаждении (криофибриноген).
Биологический смысл и санационное значение образования РФМК заключаются в том, что они способствуют поддержанию жидкого состояния крови при тромбинемии, препятствуют отложению больших масс фибрина в сосудах и уменьшают блокаду зоны микроциркуляции.
Вместе с тем доказано, что РФМК значительно легче и быстрее растворяются плазмином, чем коагулировавший фибрин.
Таким образом, свертывание крови – многоэтапный каскадный ферментный процесс, в котором последовательно активируются проферменты и действуют силы аутокатализа, функционирующие как сверху вниз, так и по механизму обратной связи. Так, первые малые дозы тромбина, чаще всего образующиеся благодаря включению внешнего механизма свертывания под влиянием тканевого тромбопластина, активируют акселераторы – факторы VIII и V, в результате чего интенсифицируется основный внутренний механизм формирования протромбиназной активности и тромбина.
Эти механизмы аутокатализа действуют интенсивно, но кратковременно. Вскоре их сменяют инактивация факторов свертывания и самоторможение системы. Этому способствуют как физиологические антикоагулянты, так и конечные и побочные продукты свертывания, многие с высокой противосвертывающей активностью. Ингибирующим влиянием на свертывание крови и тромбоцитарный гемостаз обладают и продукты фибринолиза.
По всем этим причинам свертывание крови затормаживается и не переходит в обычных условиях из локального процесса во всеобщую коагуляцию циркулирующего фибриногена.
Также в активации начальных этапов свертывания участвуют компоненты калликреин‑кининовой системы. Стимуляторами являются фактор ХIIа и его фрагменты, образующиеся в результате расщепления фактора XII калликреином. Комплекс фактор ХIIа – калликреин – высокомолекулярный кининоген (ВМК) ускоряет активацию не только фактора XI, но и фактора VII, реализуя взаимосвязь между внутренним и внешним механизмами свертывания. Еще более важно активирующее влияние тромбопластина и фактора VII на фактор IX, в результате чего даже малые дозы тканевого тромбопластина запускают процесс свертывания крови не только по внешнему, но и по внутреннему пути, через фактор IX. Установлено также, что фактор ХIIа и его фрагменты через калликреин‑кининовую систему, а отчасти и непосредственно активируют ряд других плазменных ферментных систем, в том числе фибринолитическую и систему комплемента.
Фактор VIII – многокомпонентная система, состоящая из нескольких субъединиц, участвующих в формировании его коагуляционной активности (VIII : С) и в тромбоцитарно‑сосудистом взаимодействии (фактор Виллебранда VIII : FW). Эти субъединицы отличаются разной стабильностью, гетерогенны по генетическому контролю и антигенным свойствам.
Противосвертывающие механизмы
Противосвертывающие механизмы играют ведущую роль в поддержании жидкого состояния крови и в ограничении процесса тромбообразования. Однако они изучены значительно меньше, чем процесс свертывания крови, в связи с чем вопросы функции и физиологической регуляции антикоагулянтного звена системы гемостаза во многом остаются дискуссионными.
Все образующиеся в организме антикоагулянты можно разделить на 2 группы:
1) существующие или синтезируемые самостоятельно, возникающие независимо от свертывания крови, фибринолиза или активации других ферментных систем;
2) образующиеся в процессе протеолиза – свертывания крови, фибринолиза. Химически антикоагулянты относятся к разным группам веществ – белкам, пептидам, липидам, мукополисахаридам.
Физиологические антикоагулянты, образующиеся независимо от протеолиза . К ним относятся белковые и фосфолипидные ингибиторы начальной фазы свертывания крови, из которых наиболее активен и физиологически важен относящийся к α2‑глобулинам ингибитор фактора XIа. Слабее действует на начальные фазы свертывания липидный антикоагулянт.
Из всех предшествующих антикоагулянтов наиболее активен, универсален по действию и важен для поддержания жидкого состояния крови антитромбин III (AT III). Этот белок, содержащийся в плазме в количестве около 0,3–0,42 г/л, или 3,0–4,7 ммоль/л, инактивирует не только тромбин, но и все другие активированные ферментные факторы свертывания: ХIIа, XIa, IXa, Ха. Он же является плазменным кофактором гепарина – без AT III гепарин почти совершенно не оказывает антикоагулянтного действия. Дефицит AT III, наследственный или вторичный (симптоматический), закономерно приводит к развитию тяжелейшего, часто несовместимого с жизнью тромбоэмболического синдрома. Дефицит всех других предшествующих физиологических антикоагулянтов по раздельности либо в совокупности не создает подобных критических ситуаций. Все вышеперечисленные данные закрепили за AT III репутацию главного ингибитора и модулятора системы свертывания крови. Местом синтеза AT III долгое время считали печень, однако исследования последних лет, в том числе выполненные на клеточных культурах иммунологическими методами, показали, что этот антикоагулянт продуцируется сосудистым эндотелием. Все факторы свертывания AT III инактивирует, образуя с ними эквимолярные комплексные соединения. Гепарин, соединяясь с AT III, резко ускоряет это взаимодействие и фиксирует антикоагулянт на поверхности эндотелиальных клеток, чем повышается тромбоустойчивость внутренней стенки сосудов.
Альфа2‑макроглобулин является слабым ингибитором тромбина, действие которого становится ощутимым лишь при депрессии AT III. На долю этого антикоагулянта приходится, по разным авторам, от 4 до 21% антитромбиновой активности дефибринированной плазмы. Несколько больше роль α2‑макроглобулина в связывании плазмина, но и в этом случае его действие становится ощутимым после удаления быстродействующего антиплазмина. В отличие от AT III он из всех активированных факторов свертывания взаимодействует только с тромбином. Наследственный дефицит α2‑макроглобулина не сопровождается ни сколько‑нибудь заметной тромбогенностью, ни существенными сдвигами в свертывающей системе крови, что говорит о его весьма ограниченном значении в регуляции гемостатического потенциала крови.
Более выражено ингибирующее действие на тромбин и другие активированные факторы свертывания (IXa, XIa и ХПа) α1‑антитрипсина и ингибитора 1 компонента комплемента. Однако и при их дефиците не наблюдается значительных нарушений гемостаза, что, очевидно, связано с одинаково выраженным ослаблением инактивации как свертывания крови, так и фибринолиза, вследствие чего сохраняется динамическое равновесие между этими системами.
Антикоагулянты, образующиеся в процессе свертывания крови и фибринолиза . Многие прокоагулянты и их метаболиты в процессе свертывания крови и фибринолиза приобретают антикоагулянтные свойства. Так, фибрин адсорбирует и инактивирует образующийся при свертывании тромбин, вследствие чего фибрин обозначается как антитромбин I. Эта инактивация настолько велика, что в сыворотке, как известно, остаются ничтожно малые количества тромбина. Имеются указания, что фибринопептиды, отщепляемые от фибриногена тромбином, также обладают антикоагулянтным действием.
Самоторможение наблюдается и на других этапах свертывания. Так, тромбин действует ферментативно на протромбин, отщепляя от него ингибитор фактора Ха; фактор Va после участия в свертывании начинает тормозить превращение протромбина в тромбин, а фактор ХIа после взаимодействия с фактором XII начинает тормозить его дальнейшую активацию. Мощные антикоагулянты, обладающие антитромбиновым и антиполимеразным действием, образуются в процессе фибринолиза.
Все вышеперечисленные данные в очередной раз свидетельствуют о том, что в свертывающей системе крови на всех этапах каскада действуют силы самоограничения процесса, одни и те же факторы могут выступать вначале как коагулянты, а затем – как антикоагулянты.
Схема 2 Система свертывания крови