Эпилепсия (G40)
Эпилепсия — хроническое заболевание головного мозга различной этиологии, характеризующееся повторными припадками, возникающими в результате чрезмерных нейронных разрядов и сопровождающееся разнообразными клиническими и параклиническими симптомами.
Этиология
Несмотря на многообразие этиологических факторов, а также синдромологическую неоднородность, эпилепсия остается хотя и собирательной, но единой нозологической единицей, где главным обобщающим клиническим критерием является наличие повторяющихся приступов. Одиночные или случайные эпилептические приступы не могут рассматриваться как эпилепсия, а являются, при понижении порога судорожной готовности, разновидностью реакций мозга и могут возникнуть в определенных условиях у любого человека.
Эпилептический припадок представляет собой приступ с внезапным началом, стереотипный по клиническим проявлениям, возникающий в результате нейронных разрядов, обнаруживаемых с помощью ЭЭГ, и проявляющийся в форме сенсорных, двигательных, аффективных, когнитивных и вегетативных симптомов. Наиболее важным основанием для классификации припадков является характер их начала. При генерализованных припадках приступ начинается с внезапной потери сознания и на ЭЭГ очаг не обнаруживается. Парциальные (фокальные, локальные) припадки начинаются вследствие импульса из очага (фокуса) в ограниченной части одного полушария мозга. Они подразделяются на простые и комплексные: первые отличаются от вторых отсутствием во время приступа нарушений сознания. Парциальные припадки могут распространяться и переходить в генерализованные (вторичная генерализация). Этиология заболевания может быть вызвана множеством экзогенных и эндогенных факторов. Принято считать, что основными причинами являются индивидуальная предрасположенность конституционального или наследственного характера, наличие эпилептического повреждения в мозге и локальных или генерализованных электрических изменений. При этом чем интенсивнее представлены одни из них, тем меньшая выраженность других достаточна для проявления эпилепсии. Лишь отдельные синдромы жестко детерминированы только генетическими или исключительно экзогенными причинами. Дети примерно в 4 раза чаще болеют эпилепсией, чем взрослые. Наиболее частыми причинами являются перинатальная патология и родовые травмы, врожденные пороки развития, метаболические нарушения и нарушения питания, инфекции. В среднем и пожилом возрастах в этиологии большую роль играют черепно-мозговые трамвы, сосудистые и дегенеративные заболевания мозга, опухоли. Почти в половине случаев причину эпилепсии установить не удается даже при самом тщательном обследовании. Эти идиопатические эпилепсии связаны главным образом с наследственным предрасположением, имеют, помимо других отличий, типичный возраст заболевания и в большинстве случаев хорошо реагируют на терапию. В остальных случаях припадки представляют вторичное явление по отношению к какому-либо уточненному заболеванию головного мозга. При этих симптоматических эпилепсиях (эпилептических синдромах) эндогенное предрасположение выступает в роли фактора риска. В случаях, когда при анализе особенностей клинического синдрома и данных исследования предполагается вероятность отнесения эпилепсии к симптоматической, но причины остаются невыясненными, принято говорить о криптогенной эпилепсии.
Распространенность
Распространенность эпилепсии в общей популяции составляет 7 — 10 случаев на 1000 населения. Риск развития эпилептических припадков на протяжении жизни составляет до 10 %. Заболевание может развиваться в любом возрасте, однако в 75 % эпилепсия начинается до 20-летнего возраста. Показатели заболеваемости среди мужского и женского пола практически одинаковы. Как минимум у 30 % больных со временем возникают психические расстройства и наиболее часто — при симптоматических формах.
В диагностике эпилепсии важное значение играют: семейный анамнез, возраст развития, анамнез приступов, исключение неэпилептических заболеваний, психические нарушения, эффекты проводимой терапии.
Доброкачественная детская эпилепсия с пиками на ЭЭГ в центрально-височной области («роландическая», РЭ, «сильвиева», «языковый синдром») (G 40.0).
Этиология
К настоящему времени локализованы гены, в значительной мере определяющие развитие РЭ (15ql4). Предполагаются и аутосомно-доминантное наследование с низкой пенетрантностью и возрастной зависимостью (особенно у лиц мужского пола — 60 %), и полигенное. Наследственная отягощенность весьма вариабельна (9 — 59 %). У родственников наблюдаются как аналогичные приступы, так и генерализованные.
Распространенность
РЭ относится к одной из наиболее часто встречающихся форм и составляет примерно 15–30 % всех случаев эпилепсии детского возраста; среди пациентов преобладают мальчики (в соотношении 3:2).
Клиника
Возраст начала — 3—12 лет, кульминация — в 9—10 лет.
Приступы редкие, протекают в мягкой форме и в 70–80 % случаев носят характер простых парциальных (при сохранном сознании): фаринго-оральные и односторонние лицевые миоклонии и клонии, вызывающие перекос лица, соматосенсорные ощущения (покалывания, онемение в языке, деснах, щеке с одной стороны), вокализация и остановка речи, гиперсаливация. При вторичной генерализации — гемисудороги или генерализованный припадок. Почти 75 % приступов возникает во сне, из них в 80 % — в первую половину ночи. У детей до 5 лет — преимущественно ночные, более тяжелые приступы с нарушением сознания (головокружения, боли в животе, зрительные феномены). У детей старше 5 лет — приступы более частые, но и более легкие, нередко сочетаются с приступообразными головными болями или мигренью (62 %). Примерно у 5 % больных РЭ проявляет атипичность: наряду с обычными появление других приступов (миоклонических, миоклонически-астатических, атонических, атипичных абсансов), а также более ранний возраст дебюта.
Психика и неврология, как правило, без особенностей. У 17 % детей с РЭ диагностируется нарушение внимания с гиперактивностью.
Нейропсихологическое обследование у большинства детей выявляет умеренные функциональные нарушения зрительно-моторной координации (тест Bender), снижение школьной успеваемости, дефицит внимания, памяти и поведенческие расстройства. Расстройства подобного типа, а также заикание, дислексия, энурез чаще обнаруживаются у лиц с роландическими паттернами в ЭЭГ, даже без клинических проявлений болезни.
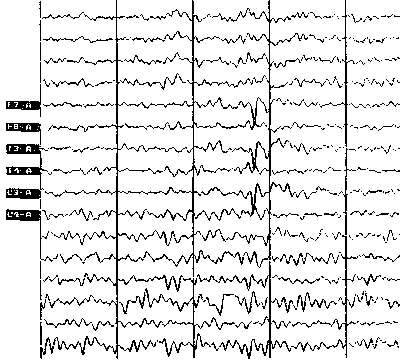
Типичная роландическая активность в левом полушарии. Скорость — 30 мм/с. Амплитуда уменьшена в 2 раза.
Диагностика
Диагноз основывается на типичных проявлениях приступов и ЭЭГ данных. На нормальном или умеренно измененном общем фоне ЭЭГ имеются локальные пики или острые волны и/или комплексы пик-волн в одном полушарии или двух, но с односторонним преобладанием в центрально-средневисочных отведениях. Характерно извращение фазы над роландической или височной областью. Эпиактивность может иногда отсутствовать, их выявлению помогает подготовка с частичной депривацией сна.
Наряду с типичными центрально-височными пиками при РЭ обнаруживаются и другие эпилептиформные паттерны. Так, примерно в 10–30 % случаев регистрируются пик-волновые комплексы, преимущественно в затылочных регионах. Морфология этих комплексов близка к роландическим и типична для другой формы эпилепсии — доброкачественной парциальной эпилепсии с затылочными пароксизмами. Частота представленности затылочных пароксизмов при РЭ обратно пропорциональна возрасту ребенка, чаще встречается до 3 лет. Примерно в 10–20 % случаев может быть зарегистрирована типичная генерализованная пик-волновая активность с частотой 3 Гц (абсансная), чаще при гипервентиляции. Пик-волновые комплексы в лобных или затылочных регионах при РЭ отмечаются примерно в 20 % случаев. Корреляции между локализацией эпилептиформной активности на ЭЭГ и особенностями течения заболевания нет.
Дифференциальная диагностика
Диффдиагностику следует проводить с оперкулярными приступами при височной эпилепсии, джексоновскими присупами. Моторными пароксизмами и центротемпоральными острыми волнами могут проявляться глиомы, каверномы, поэтому необходима нейрорадиологическая диагностика. При атипичности приступов более разнообразной пароксизмальной активности в ЭЭГ, большей выраженностью интеллектуально-мнестических и речевых нарушений дифференцировать с синдромом Леннокса — Гасто. Такой вариант РЭ получил название «атипичной РЭ», или синдрома псевдоЛеннокса.
Центрально-темпоральные спайки могут обнаруживаться у 5 % людей общей популяции здорового населения, иногда при синдромах Ретта и фрагильной Х-хромосомы, перисильвиевом синдроме (кортикальная дисплазия), каверномах, глиомах.
Прогноз
Прогноз благоприятный, в пубертатный период наступает полное выздоровление. Случаи возобновления приступов после выздоровления крайне редки (1–2 %). Факторы риска высокой частоты приступов — начало болезни с генерализованного судорожного приступа и большой временной интервал (1 год) между первым и вторым приступами. Чем раньше дебют, тем больше общая продолжительность заболевания.
Терапия
Исторически тактика терапевтических подходов при РЭ определялась дилеммой «лечить — не лечить». Определяющими факторами были, с одной стороны, абсолютно благоприятный прогноз, с другой стороны — факт наличия приступов, причем существовал явный «перекос» в оценке картины данной болезни — она рассматривалась только с точки зрения выраженности пароксизмальных проявлений. Наиболее сбалансированная точка зрения — признание необходимости лечения данной формы. Эта необходимость определяется возможностью присутствия очень частых приступов, тяжелых форм приступов, даже эпилептических статусов при РЭ. Препаратом первой очереди выбора является Султиам (Осполот). В последнее время обоснована предпочтительность применения другого антиконвульсанта (АК) — Вальпроата. Тем не менее Вальпроаты, несколько «проигрывают» Султиаму. Карбамазепин в настоящее время оттеснен на 3-е место, но реально является наиболее частым средством лечения. Барбитураты применять нежелательно. При терапии не имеет смысла добиваться улучшения ЭЭГ проявлений болезни (то есть ЭЭГ ремиссии). Лечение АК стоит проводить не более 2–3 лет клинической ремиссии, реакция болезни на отмену АК подскажет дальнейшую тактику. При лечении статуса приступов РЭ обычно применяемые препараты бензодиазепинового ряда типа диазепама малоэффективны. Особенность — предпочтительный выбор кортикостероидов (дексаметазон) в сочетании с клоназепамом.
Реабилитация: лица, ориентированные на благоприятный прогноз, становятся уравновешенными и имеют активную жизненную позицию. Там, где рекомендовались ограничения, — воспитывался комплекс неполноцености, присутствовали проблемы социальной адаптации, часто развивалось антисоциальное поведение.
Детская эпилепсия с пароксизмальной активностью на ЭЭГ в затылочной области (доброкачественная затылочная эпилепсия, ДЗЭ, эпилепсия Гасто) (G40.0).
Этиология и патогенез
ДЗЭ наследуется по аутосомно-доминантному типу с вариабельной пенетрантностью и возраст-зависимой экспрессивностью. Наличие судорожных проявлений у кровных родственников — до 37 %, мигрени — до 16 %. Это функциональная эпилепсия, развивающаяся при конституциональной эпи-предиспозиции, которая демаскирует минимальные церебральные повреждения, получаемые в родах.
Распространенность
ДЗЭ вторая по частоте форма детской идиопатической фокальной эпилепсии (10–13 %).
Клиника
Возраст начала вариабелен (15 мес. — 17 лет). Пик манифестации симптомов 5–7 лет.
Приступы и дебют имеют 2 различных варианта:
1. Ранний дебют (2–7 лет). Редкие ночные приступы, начинающиеся со рвоты, девиации глаз в сторону и нарушением сознания. Иногда — переход в гемиконвульсивный или генерализованный тонико-клонический приступ.
2. Поздний дебют (старше 7 лет). Преходящие нарушения зрения — 65 %, амавроз — 52 %, элементарные зрительные галлюцинации — 50 %, сценоподобные галлюцинации — 14 %. Сознание чаще сохранено, приступы, как правило, в дневное время. Гемиклонические судороги — 43 %, ГКТП — 13 %, автоматизмы — 13 %, версивные движения — 25 %. Послеприступное состояние в 33 % случаев сопровождается головной, чаще мигренеподобной болью, в 17 % — тошнотой и рвотой. Провоцирующие факторы: в 25 % — резкая смена освещенности при переходе из темного помещения в светлое.
Психика обычно без особенностей, иногда — эмоциональные расстройства. В нейропсихологическом статусе — снижение зрительной памяти, проявления идеомоторной апраксии.
Неврология, как правило, без особенностей.
Диагностика
Диагноз основывается на клинических данных и ЭЭГ, напоминающих таковую при роландической эпилепсии, только с другой локализацией. Локальные пики и комплексы пик-волн в одном полушарии или в двух, но с односторонним преобладанием в затылочных отведениях, которые в 38 % случаев сочетаются с генерализованными билатеральными комплексами «пик-волна», «полипик-волна». Характерно возникновение пароксизмальной активности сериями вскоре после закрывания глаз и блокирование эпилептической активности при открывании глаз. Эпилептиформная активность на ЭЭГ, а иногда и клинический приступ провоцируются фотостимуляцией. Приступная активность в ЭЭГ иногда может и отсутствовать. В то же время затылочная пик-волновая активность встречается на ЭЭГ здоровых детей с резким снижениям зрения, при синдроме Леннокса — Гасто, симптоматической затылочной эпилепсии, височной эпилепсии, при осложненной базиллярной мигрени.
Дифференциальная диагностика
Диффдиагноз следует проводить: при ранних формах — с нарушением мозгового кровообращения, при поздних формах — с симптоматической затылочной эпилепсией, парциальной эпилепсией с билатеральными затылочными кальцификатами (при целиакии, после операций на открытом сердце), митохондриальным заболеванием — синдромом MELAS, лактатацидозом, гиперглицинемией, миоклонус-эпилепсией Лафора, паразитарными заболеваниями, мигренью. При сочетании эпилепсии и мигрени важным является различие в характере галлюцинаций: для эпилепсии более характерны многокрасочные перспективные галлюцинации и сферические образы, для мигрени — чаще плоские, черно-белые, линейные. Рекомендуется проводить МР томографию во всех случаях затылочной эпилепсии.
Прогноз
При начале до 10 лет прогноз более благоприятный. Если ранний дебют, то обычно к 12 годам наступает полная ремиссия. Причиной синдрома затылочной эпилепсии с резистентностью к лечению могут быть кортикальные дисплазии. В случае синдромов, в клинической картине преобладают симптомы выпадения (амавроз, гемианопсия), а не раздражения (фотопсии).
Терапия
Средство первого выбора — Вальпроат, в странах Европы — Султиам (Осполот), Ламиктал, Карбамазепин. Средства второго выбора — Бензодиазепины (Клобазам), комбинация обоих препаратов.
Локализованная (фокальная, парциальная) симптоматическая эпилепсия и эпилептические синдромы с простыми парциальными припадками (G40.1).
В Международной классификации эпилепсии к рубрикам МКБ— 10 G40.1 и G40.2 отнесены локализационно обусловленные симптоматические формы с известной этиологией и верифицированными морфологическими нарушениями.
Приступы без нарушения сознания
Эта группа простых парциальных припадков включает моторные, вегетативные приступы и разнообразные сенсорные и соматосенсорные припадки, во время которых сознание не нарушается.
Простые парциальные приступы, которые переходят во вторично генерализованные приступы
Критерий генерализации — выключение (а не изменение) сознания. Припадки могут быть судорожными и бессудорожными. Кроме нарушения сознания характерно: а) массивные вегетативные проявления; б) двусторонние синхронные и симметричные разряды на ЭЭГ.
Парциальный припадок может переходить в комплексный (сложнопарциальный).
Локализованная (фокальная, парциальная) симптоматическая эпилепсия и эпилептические синдромы с комплексными парциальными судорожными припадками (G40.2).
Комплексные парциальные припадки заменили ранее употребляемые термины «психомоторные припадки» и «височная эпилепсия». Им часто предшествуют простые парциальные приступы. При этих припадках нарушена способность осознания происходящего или адекватного ответа на стимулы. Практически обязательная особенность сложнопарциального приступа — это симптомы нарушения когнитивных функций. Чаще — идеаторные (навязчивая, странная, ненужная мысль) — то есть форсированное мышление, иллюзии восприятия времени и симптомы дереализации-деперсонализации, например, «уже виденного». Дисмнестические феномены, например, насильственные воспоминания, носят характер экмнезий.
Эпилепсия лобной доли (фронтальные эпилепсии, ФЭ) (G40.1/G40.2).
Этиология и патогенез
Часто обнаруживается этиологическая связь с очаговой атрофией, травмами, нейроинфекциями, опухолями (астроцитомы и олигодендроглиомы) или артерио-венозными мальформациями (АВМ). Нередко причиной является обнаруживаемые с помощью ЯМР нарушения миграции нейронов или дисгенезии. Эпилептический статус формируется при эпилепсии лобной доли особенно часто.
Распространенность
Среди симптоматических форм составляет 15–20 %.
Клиника
Возраст начала — любой.
Приступы обычно частые, с нерегулярными интервалами, нестереотипные, часто во время сна. Нередки автоматизмы жестов с внезапным началом и окончанием, почти без постприпадочной спутанности, продолжительностью обычно менее 30 с, эмоционально окрашенные речевые автоматизмы. Автоматизмы часто причудливы, бурные («двигательная буря»), сексуально окрашены, истероподобны. Редко неопределенная аура или парциальный соматосенсорный припадок в виде ощущения тепла, дуновения, паутины, мягкого прикосновения.
Приступы при эпилепсии дополнительной моторной зоны (префронтальная) проявляются в виде постуральных, простых фокальных тонических с вокализацией, позой фехтовальщика, остановкой речи, размахиванием руками либо сложные фокальные с недержанием мочи.
Приступы при цингулярной эпилепсии (поясная извилина) — это комплексные фокальные припадки с начальными автоматизмами сексуального характера, вегетативными проявлениями, изменениями настроения, возбуждением, недержанием мочи.
Для приступов с очагом в передней (полюс) лобной области характерны насильственное мышление, вегетативное сопровождение, утрата реактивности — «псевдоабсанс». Припадки начинаются с потери контакта, адверсивного и вслед за этим контраверсивного движения глаз и головы, аксиальных клонических подергиваний, падения, а также с автономных проявлений. Очень часто переходят в генерализованные тонико-клонические судороги.
Припадки орбито-фронтальной области являются комплексными фокальными; сначала появляются проявления автоматизма или обонятельные галлюцинации, вегетативная пароксизмальная симптоматика и мочеиспускание.
Припадки дорсолатеральные являются простыми фокальными тоническими (вращения, пропульсии, поклоны), сопровождаются афазией и комплексными фокальными с начальными автоматизмами, без ауры.
Приступы оперкулярной эпилепсии парциальные с клониями в лице, эпигастральными ощущениями, вкусовыми галлюцинациями, торможением речи, страхом и вегетативными симптомами. Сложные парциальные припадки с глотательными, жевательными движениями, слюнотечением, лярингеальными симптомами.
При лобной моторно-кортикальной эпилепсии — парциальные джексоновские припадки с послеприпадочным параличем Тодда. При вовлечении прероландической коры — остановка речи, вокализация, афазия.
Психика: «лобные» изменения личности, эксцентричность, персеверативное и инертное поведение, трудности социальной адаптации, расторможенность, снижение критики. При дорсолатеральной эпилепсии психика достаточно быстро изменяется, наблюдаются персеверация, расторможенность, ухудшаются когнитивные процессы.
Неврология соответствует этиологическому фактору (опухоль, локальные лобные деструктивные нарушения при травме).
Диагностика
Исходит из этиологических факторов, клинических типов припадков, психических и неврологических особенностей, нейрорадиологической диагностики, КТ, ЯМР, ангиографии и ЭЭГ данных.
ЭЭГ при эпилепсии лобной доли часто оказывает лишь незначительную помощь. Иктальная ЭЭГ показывает уплощение ритмических полиспайков (16–24 /с) и вторичную генерализацию из очага. При цингулярной эпилепсии точная локализация очага возможна только по СЭЭГ (стереотаксическая ЭЭГ). ЭЭГ орбито-фронтальной области во время припадка сглажена, с появлением ритмичных полиспайков 16–24 /с и вторичной генерализацией. При дорсолатеральной эпилепсии в большинстве случаев очаг можно хорошо определить регистрацией поверхностной ЭЭГ во время припадка или интериктальном периоде. ЭЭГ при лобной моторно-кортикальной эпилепсии в 75 % случаев без фокальной патологии.
Дифференциальная диагностика
Детальное обследование больных с лобной эпилепсией позволяет в первую очередь исключить текущие церебральные процессы.
Общими особенностями пароксизмов при эпилепсии с очагом в лобной доле является типичная феноменология приступов: тоническая или постуральная активность, повышенная двигательная активность, комплексные жестикуляционные автоматизмы, вокализации, их частота, кратковременность, отсутствие или незначительная спутанность сознания после приступа. Последнее часто приводит к ошибочной трактовке припадка, как психогенного. Отличать припадки лобной доли от психогенных очень сложно, и в первую очередь в связи с тем, что нередко оба вида припадков могут наблюдаться у одного и того же больного. В большинстве диагностически сложных случаев окончательный диагноз может быть поставлен только после видео- и телеэнцефалографического мониторирования. Приступы лобной доли можно принять за припадки, исходящие из височной доли головного мозга. Нередко затруднения возникают в дифференциальной диагностике эпилептических вегетативно-висцеральных припадков и обмороков, относящихся к аноксическим (аноксоишемическим) припадкам.
Прогноз
Течение ФЭ характеризуется нередко неблагоприятными тенденциями и более прогредиентно при преобладании более ранних экзогенных факторов в этиологии, начале заболевания с частых припадков, наличии грубых психопатологических расстройств и изменений на ЭЭГ органического типа. Прогноз зависит также от локализации очага в лобной доле.
Терапия
Фронтальные эпилепсии относятся к трудным для терапии формам. Средства первого выбора АК — Карбамазепин. Второй выбор — Вальпроат, Дифенин, Гексамидин. Этиологическая (симптоматическая) терапия. При неэффективности — хирургическое лечение.
Эпилепсия височной доли (височная эпилепсия, ВЭ).
Этиология
Причинами ВЭ являются перинатальная травма и гипоксемия, посттравматический очаговый глиоз височного полюса, склероз гиппокампа, постэнцефалитические изменения, травма, ганглиоглиомы, малые глиомы, АВМ, венозные ангиомы и рубцы после мозговых инфарктов, церебрально-сосудистые нарушения в позднем возрасте.
Распространенность
ВЭ чаще всего встречающаяся форма симтоматической локализованной эпилепсии (60–65 %).
Клиника
Возраст начала — любой, но чаще или в детстве или во втором десятилетии жизни.
Приступы: элементарно-фокальные (обонятельные, слуховые, эпигастральные феномены), комплексные парциальные, вторично генерализованные. Комплексные парциальные часто начинаются с остановки движения с оро-алиментарными автоматизмами. Длительность более минуты, нечеткое окончание, послеприступная спутанность, амнезия приступа. Приступы часто серийные.
При гиппокампальной (медиобазальная лимбическая или первичная ринэнцефалическая психомоторная) форме, составляющей 70–80 % эпилепсии височной доли, припадки появляются в группах или по отдельности: бывают комплексными очаговыми, начинающиеся со странных неописуемых ощущений, галлюцинаций или иллюзий с последующим отключением (оцепенением взгляда), ротаторными или пищевыми автоматизмами. Продолжаются в среднем 2 минуты. При прогрессировании могут отмечаться генерализованные тонико-клонические судороги.
Амигдалярная эпилепсия (передняя полюсно-амигдалярная) сопровождается припадками с эпигастральным дискомфортом, тошнотой, выраженными вегетативными симптомами и другими проявлениями (отрыжка, бледность, отечность, покраснение лица, диспноэ, мидриаз, страх, паника, обонятельно-вкусовые галлюцинации). Ступор, бессознательное состояние наступают постепенно, сопровождаются оцепеневшим взглядом, оральными и пищевыми автоматизмами, внешними проявлениями «растерянности». Сочетание с генерализованными тонико-клоническими судорогами, имеющими фокальное начало, является редким (30 %).
При латеральной задневисочной эпилепсии припадки с аурой в виде слуховых галлюцинаций, зрительных галлюцинаций с нарушением речи в случае локализации очага в гемисфере, доминантной для речи. Вслед за этим наступают дисфазия, нарушения ориентировки или продолжительные слуховые галлюцинации, движения головы в одну сторону, иногда автоматизмы с остановкой взгляда. Часто — сноподобные состояния (Dreamy State).
Оперкулярные (инсулярные) эпилепсии проявляются вестибулярными или слуховыми галлюцинациями, отрыжкой или вегетативными проявлениями, односторонними подергиваниями лица и парестезиями. Бывают обонятельно-вкусовые галлюцинации.
Психика: часто трудности обучения, нарушения памяти, тенденция к персеверациям, эгоцентризм, обстоятельность, аккуратность, повышенное чувство долга, конфликтность, эмоциональная лабильность.
Неврология: зависит от этиологии, часто скудна.
Диагностика
Исходит из этиологических факторов, клинических типов припадков, психических и неврологических особенностей, нейрорадиологической диагностики, КТ, ЯМР, ангиографии и ЭЭГ данных. ЭЭГ между припадками показывает типичные передневисочные острые волны, особенно при регистрации во сне. Для ЭЭГ, снятой во время припадка, типично начальное одностороннее уплощение, особенно на височных отведениях. При СЭЭГ регистрируются высокочастотные (16–28 Гц) пики низкого вольтажа, исходящие из одного гиппокампа и распространяющиеся в миндалевидное тело и поясную извилину того же полушария или медиобазальные структуры контралатеральной стороны. При приступах по типу автоматизмов — возможна ритмичная первично- или вторичногенерализованная тета-активность без острых феноменов.
Дифференциальная диагностика
В диффдиагностике следует учитывать, что височные приступы можно принять за приступы лобной эпилепсии. Объем специальных исследований и проведение дифференциального диагноза — как и при парциальных симптоматических эпилепсиях лобной доли.
Прогноз
Течение ВЭ характеризуется неблагоприятными тенденциями и более прогредиентно при преобладании ранних экзогенных факторов в этиологии, начале заболевания с частых припадков, наличии грубых психопатологических расстройств и изменений на ЭЭГ органического типа. Прогностически благоприятными являются упрощение припадков, переход от сложных форм парциальных припадков к простым, а при судорожных проявлениях — от развернутых к абортивным. У 30–40 % при правильной терапии можно добиться прекращения приступов, стойких медикаментозных ремиссий.
Терапия
Медикаментозное лечение симптоматичеких фокальных эпилепсии в большинстве случаев является сложным. Средства первого выбора АК — Карбамазепин. Второй выбор — Вальпроат, Дифенин, Гексамидин. Этиологическая (симптоматическая) терапия. При неэффективности — хирургическое лечение.
Эпилепсии затылочной и теменной доли (затылочные и теменные эпилепсии, ЗЭ, ТЭ).
Этиология
При ТЭ наиболее часто встречаются в этиологии нейроинфекции и черепно-мозговые травмы, опухоли и артериовенозные аневризмы. Вместе с тем приступы ТЭ могут являться следствием резидуального мозгового поражения. При ЗЭ преобладают любые деструктивные корковые нарушения; в пожилом возрасте — чаще опухоль, последствия НМК.
Распространенность
ТЭ встречается значительно чаще, чем ЗЭ.
Клиника
Возраст начала любой. Большинство больных с ТЭ к началу заболевания были старше 16 лет. Заболевание редко начинается до 6 лет. Эта особенность отличает ТЭ от ЗЭ.
Припадки при ТЭ представляют собой простые парциальные сенсорные приступы в виде ощущений покалывания, онемения, с ощущением электризации. Парестезии могут быть ограниченными или распространяться по типу джексоновских. Может возникнуть желание перемещения части тела или ощущение, как будто часть тела уже двигалась. Чаще всего поражаются те участки, которым соответствует наибольшая площадь коркового представительства — например, рука, плечо и лицо. Могут возникать ощущения онемения с покалыванием языка, жесткого или холодного языка. Сенсорные нарушения в области лица могут быть двусторонними. Иногда, особенно при поражении нижней и латеральной париетальных долек, появляется ощущения тошноты, захлебывания или удушья. Ощущение боли возникает редко, воспринимается чаще как поверхностное жжение или эпизодически возникающее, неопределенное очень болезненное ощущение.
Зрительные проявления поражения париетальной доли могут быть красочными и приобретать звериный вид. Может возникать метаморфопсия с искажением, сокращением или удлинением образа, которая чаще наблюдается при разрядах в недоминантном полушарии. Наряду с этими «положительными» феноменами или продуктивной симптоматикой образуются и так называемые негативные феномены, проявляющиеся, кроме онемения, ощущением отсутствия какой-либо части тела, утратой способности осознавать часть или половину тела — асоматогнозия (чаще при правосторонних припадках). Тяжелое головокружение может свидетельствовать о вовлечении супрасильвиевой париетальной доли. Припадки левой задней доли сопровождаются рецептивными и кондуктивными нарушениями речи (центр Вернике).
Довольно редко встречающееся сенсорное нарушение с участием парацентральной дольки охватывает обе нижние конечности. Припадки парацентральной дольки имеют тенденцию к вторичной генерализации.
При ЗЭ обычно припадки проявляются зрительными симптомами: простыми — летучие зрительные порропсии (скотома, гемианопсия, амавроз, или искры, вспышки). Чаще они в поле зрения, противоположном месту разряда в зрительной коре. Иллюзии восприятия с искажением предметов: односторонняя диплопия, изменения размера, расстояния, расположение объектов в определенной части пространства, искажение предметов, внезапное изменение формы. Зрительные галлюцинаторные припадки могут быть комплексными и принимать вид красочных сцен. Наряду с этим сцена может быть искажена или уменьшена, иногда человек может увидеть свое собственное изображение (разряды в височно-затылочной коре).
Припадки могут проявляться без зрительных симптомов — контраверсией глаз или головы и глаз, подергиванием век, насильственным закрыванием глаз, ощущением дрожания глаз или всего тела, головокружением изолированным или головокружением и шаткой ходьбой, вместе с головной болью и мигренью.
Неврология — очаговая неврологическая симтоматика.
Диагностика
Исходит из этиологических факторов, клинических типов припадков, психических и неврологических особенностей, нейрорадиологической диагностики, ангиографии и ЭЭГ данных. ЭЭГ при эпилепсии париетальной доли сопровождается соответствующим образом локализованными разрядами острых волн. При ЭЭГ ЗЭ разряды фокальные, могут распространяться в височную долю (тогда присоединяются симптомы задневисочного, гиппокампального или амигдалярного припадка). Если первичный фокус находится в area supracalcarinea — разряд может распространяться на супрасильвиеву зону и приобретать симптоматику приступов париетальной доли или дополнительной моторной зоны.
Дифференциальная диагностика
Следует учитывать, что данные формы эпилепсии как по клиническим проявлениям, так и по специфическим изменениям ЭЭГ значительно труднее в дифференциальной диагностике, чем при дифференциации других парциальных эпилепсии. Объем специальных исследований и проведение дифференциального диагноза — как и при других парциальных симптоматических эпилепсиях.
Прогноз
Зависит от прогредиентности этиологического фактора. Течение имеет три основных типа: с быстрым нарастанием частоты и тяжести приступов, стабильный тип с относительным постоянством приступов и доброкачественный с постепенным урежением и ослаблением приступов.
Терапия
Медикаментозное лечение симптоматических фокальных эпилепсии в большинстве случаев является сложным. Средства первого выбора АК — Карбамазепин. Второй выбор — Вальпроат, Дифенин, Гексамидин. Этиологическая (симптоматическая) терапия. При неэффективности — хирургическое лечение.
Генерализованная идиопатическая эпилепсия и эпилептические синдромы (G40.3).
Генерализованные идиопатические эпилепсии (связанные с возрастом) — это те формы, припадки которых всегда вначале генерализованы, а их проявлением на ЭЭГ является нормальная активность фона и только пароксизмальные Генерализованные двусторонние симметричные разряды, которые увеличиваются в период медленного сна. Приступы появляются обычно на фоне совершенно нормального состояния. Больные не имеют локальных ЭЭГ и других нейрорадиологических изменений.
Генерализованные идиопатические и/или симптоматические эпилепсии включают в себя формы как идиопатических, так и симптоматических вариантов (таких как Уэста и Леннокса — Гасто, хотя они и вынесены в МКБ-10 в отдельную рубрику — G40.4), а также таких, статус которых не определен. Некоторые их признаки свидетельствуют об идиопатическом происхождении (сильная генетическая предрасположенность, отсутствие известной этиологии), некоторые — о симптоматическом происхождении (неврологические изменения, задержка умственного развития).
Генерализованные симптоматические эпилепсии встречаются в основном в грудном возрасте и детстве. Эти генерализованные припадки в самом деле чем-то отличаются от идиопатических, также как и их ЭЭГ. Они чаще не однотипны, а включают и миоклонические подергивания, и тонические приступы, и атонические, и атипичные изменения сознания. ЭЭГ изменения, конечно, тоже двусторонние, но менее регулярные, менее правильные и механические, чем при идиопатических формах, а также всегда более или менее асимметричны.
Главное в ЭЭГ при всех этих формах — это то, что в межприступный период они тоже патологичны — это супрессивные вспышки, медленные пик-волны, генерализованные быстрые ритмы или даже гипсаритмия. Частые находки и очаговые непароксизмальные изменения. Однако самое главное — присутствуют клинические, нейрорадиологические и, конечно, нейропсихологические признаки энцефалопатии, специфической или неспецифической.
Доброкачественные: миоклоническая эпилепсия раннего детского возраста (доброкачественная миоклоническая эпилепсия младенческого возраста).
Этиология
Относится к идиопатическим формам. Этиология неизвестна, но часто имеется семейный анамнез судорог или эпилепсии.
Распространенность
Очень редко, до 70 % — мальчики.
Клиника
Возраст начала чаще в 1–2 года.
Припадки носят характер очень кратковременных генерализованных миоклоний.
Психика чаще без изменений, может наблюдаться некоторое запаздание интеллектуального развития.
Неврология — без особенностей.
Диагностика
Основой для диагностики являются характер приступов, этиологические факторы и данные ЭЭГ, которые, как правило, в пределах нормы или имеют умеренные изменения, иногда с острыми волнами, пиками, комплексами пик-волн, острая-медленная волна, преобладающими в ранних стадиях сна. ЭЭГ во время припадка — картина генерализованной эпилептической активности с нерегулярными пиками, пик-волнами, острыми волнами, обычно асимметричными, иногда — билатерально-синхронными.
Дифференциальная диагностика
Миоклонии детского возраста всегда составляют значительные сложности в дифференциально-диагностическом плане, так как нередко встречаются даже в норме. Физиологические миоклонии наблюдаются во сне практически у всех здоровых людей.
Патологические миоклонии делятся на эпилептические и неэпилептические; последние возникают при разнообразных заболеваниях и связаны с поражением серотонинергических нейронов ядра шва или берущих отсюда начало восходящих и нисходящих путей, а также при поражении зубчатого ядра мозжечка и передних его ножек. Миоклонии могут иметь гередитарное происхождение (например, при фенилкетонурии, мозжечковой диссинергии), а также быть результатом энцефалопатий (постаноксической, интоксикационной, дисметаболической) и при тяжелых энцефалопатиях типа парамиоклонуса Фридрейха, миоклонической церебральной диссинергии Ханта. Миоклонические судороги типичны при ревматическом энцефалите, болезни Крейцфельда — Якоба, склерозирующем подостром лейкоэнцефалите и др.
Эпилептические миоклонии позволяет дифференцировать ЭЭГ.
Важным в диагностике является учет возраста начала заболевания, что облегчает диффдиагностку с синдромом Леннокса — Гасто, эпилепсией Янца, миоклонической эпилепсией Унферрихта — Лундборга. На начальных этапах особые сложности возникают при разграничении с синдромом Уэста.
Прогноз
Как правило, очень благоприятный. Приступы хорошо реагируют на лечение, но в подростковом возрасте могут встречаться ГТКП (генерализованные тонико-клонические приступы).
Терапия
Вальпроаты (депакин, орфирил и др.).
Неонатальные приступы (семейные) (доброкачественные семейные идиопатические неонатальные судороги).
Этиология
Доказан аутосомно-доминантный тип наследования. Ген картирован на длинном плече 20-й хромосомы, локус 20q13.2, второй ген на длинном плече 8-й хромосомы, локус 8q24.
Распространенность
Относятся к редким формам эпилепсии, к настоящему времени описано менее 200 случаев. Одинаково часто у мальчиков и девочек, в 100 % случаев — наследственное отягощение с аналогичными приступами в период новорожденности.
Клиника
Возраст начала — 1—7-й день жизни, максимально часто 2—3-й сутки.
Приступы достигают частоты до 3–6 в день, длительностью 1–8 минут. Судороги сцеплены с ритмом «сон-бодрствование», чаще во сне. Приступы носят чаще фокальный характер: мягко протекающие кратковременные припадки типа апноэ или клоний, тонических проявлений, характерны глазные симптомы (фиксация взгляда широко раскрытых глаз, девиация глаз, вверх, нистагмоидные подергивания, моргания век, расширение зрачка), ороавтоматизмы (орофациальные, ороалиментарные). Период персистирования приступов — до нескольких недель.
Психика — без особенностей.
Неврология — без особенностей.
Диагностика
Основой для диагностики являются характер приступов, уточненные этиологические факторы и данные ЭЭГ, хотя они, естественно, недостаточно изучены. Очень низкая амплитуда активности в этом возрасте сравнима с уровнем «шума» самого энцефалографа. Специфических феноменов межприступная ЭЭГ не содержит. Во время приступа — билатеральная симметричная супрессия амплитуды на 5— 19 с (тоническая фаза с апноэ), затем — ритмичные вспышки «крутых» медленных волн, прерывающиеся высокоамплитудными полиспайками и острыми волнами (клоническая фаза).
Дифференциальная диагностика
Проводится с метаболическими нарушениями, перинатальными повреждениями и аномалиями головного мозга, доброкачественными идиопатическими судорогами новорожденных («судороги пятого дня»), инфекциями, недостаточностью холекальциферола.
Прогноз
Приступы спонтанно прекращаются через несколько недель жизни (68 % — в первые 6 недель), без последствий. У части детей повторно возникают судороги в 3–4 мес. жизни, 10–15 % трансформируются в эпилепсию.
Терапия
Фенобарбитал 5 мг/кг/сут.
Детский эпилептический абсанс (пикнолепсия) (абсансная эпилепсия Кальпа).
Этиология
Выраженная генетическая предрасположенность у нормальных во всех других отношениях детей. Чаще встречается у девочек — примерно в 1,5–2 раза.
Распространенность
Абсансы — один из наиболее частых типов приступа у детей и подростков, ежегодно впервые диагностируются у 6 — 13 детей на 100 000 детского населения (до 16 лет). Абсансы составляют до 50 % случаев всех генерализованных форм эпилепсии. Пикнолепсия составляет 8 — 10 % всех эпилепсии.
Клиника
Возраст начала 5–8—10 лет.
Приступы — простые (типичные и атипичные) абсансы, обычно серийные и могут быть чрезвычайно частыми — до 50 и более в день. Начинаются внезапно, без предвестников, кратковременным выключением сознания и также внезапно заканчиваются. После припадка не остается никаких следов психических нарушений, больные продолжают начатую деятельность. Простые абсансы встречаются примерно в четверти случаев, длятся секунды с так называемым «пустым взглядом», нередко направленным вверх. Нередко приступы сопровождаются частым миганием век, ретропульсией головы (сложные абсансы). Сложные абсансы более характерны и сопровождаются минимальным моторным (тоническим — 50 %, миоклоническим — 38 %, атоническим — в единичных случаях, с автоматизмом — 37 %), а также вегетативным компонентом (5 %) и даже фокальными феноменами (12 %). У одного пациента могут наблюдаться различные виды абсансов. Примерно у трети больных абсансы сочетаются с большими генерализованными судорожными припадками во время бодрствования. В 7—24 % случаев при пикнолепсии развивается статус абсансов (пик-волновой ступор).
Приступы могут провоцироваться напряженной умственной работой или, наоборот, состоянием «скуки», депривацией сна, фотостимуляцией, почти в 100 % случаев — гипервентиляцией. Нарушение когнитивных функций может быть результатом неправильного лечения (барбитураты).
Психика: интеллектуальный дефицит наблюдается не более чем у 5 % детей с пикнолепсией и чаще всего при атипичных абсансах. Примерно в 25 % случаев имеются гиперактивность и дефицит внимания.
Неврологический статус, как правило, нормальный.
Диагностика
Основой для диагностики являются характер приступов, уточненные этиологические факторы и данные ЭЭГ.
По ЭЭГ данным частота выявляемости типичной абсансной активности в межприступном периоде — до 85 %. Наиболее типичный паттерн — вспышки генерализованной высокоамплитудной пик-волновой активности с частотой 3 в секунду. Характерно внезапное возникновение разряда и более плавное прекращение. Гипервентиляция легко провоцирует пароксизмальную активность и служит хорошим критерием адекватности терапии.
Дифференциальная диагностика
Проводится с другими формами эпилепсии, сопровождающимися абсансами. Ювенильный эпилептический абсанс позволяет дифференцировать возрастной аспект начала приступов. При сложных абсансах — с эпилепсией с миоклоническими приступами. Важную роль в дифференциальной диагностике играют данные ЭЭГ — корреляция приступов с типичной картиной абсансной активности.
Прогноз
Считавшиеся благоприятными течение и исход пикнолепсии к настоящему времени рассматриваются иначе. Утверждение, что приступы исчезают ближе к периоду половой зрелости, подтверждается лишь в 60 % случаев. У части больных припадки лишь урежаются, принимают иную форму или присоединяются генерализованные судорожные припадки. Полная терапевтическая ремиссия достигается в 80 % случаев. При раннем начале лечения и адекватной терапии прогноз более благоприятен.
Терапия
В настоящее время препарат выбора — Вальпроат. Применение этосуксимида (суксилеп) в монотерапии не предотвращает развитие ГТКП. Возможна комбинация этих препаратов. Сочетание суксилепа с барбитуратами резко увеличивает частоту когнитивных и поведенческих расстройств. Карбамазепин обычно способствует учащению абсансов, и в связи с этим применяется иногда как своеобразный тест с диагностической целью. Применение барбитуратов приводит к развитию резистентности абсансов к другим базовым препаратам.
Лечение следует проводить до пубертатного периода.
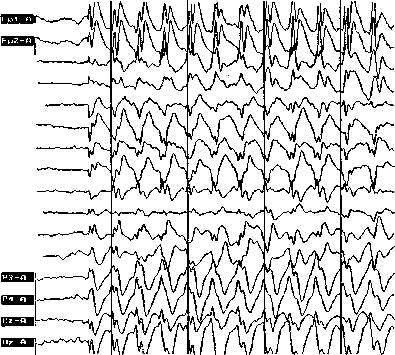
Типичный абсанс — первично генерализованный разряд высокоамплитудных комплексов «пик-волна» с частотой 3 в секунду. (Уменьшено в 4 раза.). Ребенок 7 лет
Эпилепсия с большими судорожными приступами Grand mal во время пробуждения.
Этиология
Вероятно, это форма вытекает из нелеченных или недолеченных пикнолепсий детской и ювенильной. Эпилептическая система при этом изменяется, поэтому в каждом случае приходится ее уточнять и подбирать медикамент индивидуально. В связи с малой проявляемостью этих эпилепсии на ЭЭГ, целесообразно уточнение эпилептической системы нейропсихологическими методами. Генетическая предрасположенность довольно четкая; от 4 до 12 % членов семьи страдают эпилептическими приступами.
Распространенность
Приблизительно 25 % всех эпилепсии с «большими припадками» следует относить к данному синдрому.
Клиника
Начало — синдром чаще развивается на 2-м десятилетии жизни, преимущественно в период полового созревания.
В подавляющем большинстве случаев припадки ГТКП (генерализованный тонико-клонический припадок) возникают вскоре после пробуждения (90 %), независимо от времени пробуждения. Второй суточный пик припадков — в вечернее время, в релаксации. Существует выраженная корреляция с повышенной светочувствительностью.
Приступы — обычный большой развернутый припадок сразу после пробуждения или в течение 1–1,5 часа после пробуждения. Если имеется при этом другой тип припадков, то это скорее всего абсансы или миоклонические припадки.
Диагностика
Основывается на типичности приступов и времени их возникновения. ЭЭГ уточнение проблематично, так как приступы скорее будут регистрироваться в «фазовых» состояниях, когда человек «не до конца проснулся». Помогает депривация сна. До 30 % пациентов выявляют светочувствительность.
Прогноз
Даже при устойчивой терапевтической ремиссии припадков прекращение лечения следует начинать не ранее чем спустя пять лет после исчезновения приступов при наличии хороших результатов на ЭЭГ, а также после достижения двадцатилетнего возраста.
Терапия
Достаточный и регулярный сон, соблюдение режима сна-бодрствования имеет важное значение. На первом месте — вальпроаты, на втором — фенитоины. Применение карбамазепинов противопоказано, так как при этой форме нередки абсансы и миоклонические приступы.
Ювенильная миоклоническая эпилепсия (эпилепсия с импульсивными Petit Mal, ЮМЭ, с миоклоническим Petit Mal, синдром Янца, синдром Герпина — Янца).
Этиология
Форма генерализованной идиопатической эпилепсии с выраженным генетическим предрасположением, идентифицированным генетическим дефектом (короткое плечо 6-й хромосомы на расстоянии 21 сМ от теломеры и локус 15ql4).
Распространенность
Синдром частый, составляет около трети случаев с дебютом в подростковом возрасте и до 11–12 % среди всех форм эпилепсии.
Клиника
Возраст начала: 12–20 лет.
Приступы короткие, «простреливающие», билатерально-синхронные, массивные, симметричные миоклонии, преимущественно в руках и верхнем плечевом поясе, в большинстве случаев с сохраненным сознанием. При вовлечении ног — внезапное падение. Иногда припадки следуют залпами. Как правило, после пробуждения при движении, провоцируются бессонницей. Обычно комбинируются с генерализованными тонико-клоническими приступами, которые возникают или при пробуждении, или вечером в состоянии расслабления («эпилепсия конца рабочего дня»).
Неврология без особенностей, иногда — фокальная микросимптоматика, оживление глубоких рефлексов.
Психика: характерологические особенности по типу непостоянства, поверхностности, недостаточной критичности, недооценки заболевания.
Диагностика
Основывается на типичных клинических проявлениях. На ЭЭГ обычно хорошо выраженный и широко распространенный альфа-ритм, иногда заостренные волны или комплексы множественных пиков или множественные пик-волны. Нет непосредственной корреляции между ЭЭГ пиками и подергиваниями. Часто наблюдается повышенная светочувствительность.
Дифференциальная диагностика
Эпилептические миоклонии возникают при разнообразных заболеваниях, о чем указано в разделе диффдиагностики миоклонической эпилепсии детского возраста.
Важным в диагностике является учет данных ЭЭГ, возраста начала заболевания, что облегчает диффдиагностку с доброкачественной миоклонической эпилепсией детского возраста, синдромом Леннокса — Гасто, миоклонической эпилепсией Унферрихта — Лундборга, синдромом Уэста.
Прогноз
При адекватной терапии и отрегулируемым образом жизни прогноз благоприятный. Приступы могут персистировать в зрелом возрасте «большими припадками». Почти у всех больных после отмены лечения приступы возобновляются, поэтому даже при многолетнем отсутствии припадков нельзя прекращать прием антиконвульсантов. Социальный и витальный прогнозы благоприятные.
Терапия
Первый выбор — Вальпроат. Второй выбор — Этосуксимид, Клоназепам, Гексамидин.
Эпилепсия с миоклоническим абсансом (синдром Тассинари) (G40.4).
Этиология
Данный синдром относится к криптогенным формам эпилепсии.
Распространенность
Встречается крайне редко, в основном у мальчиков.
Клиника
Возраст начала в 4–9 лет, в среднем — 7 лет.
Приступы клинически характеризуются нарушением сознания по типу абсансов, которые сопровождаются тяжелыми двусторонними ритмическими клоническими (абсансы с миоклониями плечевого пояса) подергиваниями, часто сочетающимися с тоническими сокращениями. Припадки наблюдаются несколько раз в день, осознавание подергиваний может быть сохранено. Сочетанные припадки бывают редкими.
Неврология: без грубых органических нарушений.
Психика: в основном психомоторное развитие соответствует возрасту, но с развитием заболевания возможно отставание.
Диагностика
ЭЭГ: всегда двусторонние синхронные и симметричные разряды ритмических пик-волн 3 Гц, так же как и при типичных абсансах.
Дифференциальная диагностика
Проводится с другими формами эпилепсии, сопровождающимися абсансами. Основную роль в диффдиагностике играют данные ЭЭГ — корреляция приступов с типичной картиной абсансной активности. Учитывая, что типичные абсансы наблюдаются почти исключительно в детском возрасте, важным для диагностики является возраст их начала.
Прогноз
В отношении приступов и психического развития является куда менее благоприятным, чем при пикнолепсии, в связи с довольно выраженной резистентностью припадков к терапии, умственным отставанием и возможным переходом в другие виды эпилепсии, например в синдром Леннокса — Гасто. Нередко миоклонии вообще не поддаются лечению.
Терапия
Первый выбор: Этосуксимид, Вальпроат. Второй выбор — Карбамазепин, Бензодиазепины. Рекомендуется сочетание Вальпроата и Этосуксимида, Вальпроата и Ламотриджина, Вальпроата и Клобазама, применение новых генераций АК — Фелбамат, Габапентин, Вигабатрин.
Эпилепсия с миоклонически-астатическими приступами.
Этиология
Часто генетическая предрасположенность. У 37 % больных выявлено семейное заболевание этой формой.
Распространенность
Встречается редко, мальчики поражаются чаще, чем девочки.
Клиника
Возраст начала между 7 мес. и 6-м годом жизни, обычно 2–5 лет.
Приступы на фоне правильного психомоторного развития начинаются обычно с фебрильных или афебрильных тонико-клонических приступов, малых атонических, миоклонических, миоклонически-астатических припадков и сложных абсансов. Часто припадки идут в виде статусоподобных серий. Бессудорожные приступы составляют 36 % всех случаев. В приступе бывает и тонический компонент, и даже чистые тонические приступы, но они возникают на поздних стадиях заболевания, и обычно в неблагоприятных ситуациях. Это отличает данную форму от синдрома Уэста, которому они как раз свойственны.
Неврология: обычно без грубых органических нарушений.
Психика: в 50 % случаев психомоторное развитие соответствует возрасту.
Морфология: без структурных нарушений.
Диагностика
Проводится с учетом этиологии, клиники приступов. ЭЭГ в начале болезни — без особенностей или с преобладанием тета-ритма. С развитием болезни — на нормальном или умеренно измененном фоне, нерегулярные комплексы пик-волна и полипик-волна 3–4 Гц. Может напоминать картину при синдроме Леннокса — Гасто, но с менее выраженной дезорганизацией и тенденцией к генерализованным, регулярным комплексам пик-волна. Выражена фотосензитивность. Фокальные и мультифокальные проявления обычно отсутствуют.
Дифференциальная диагностика
При неэффективности терапии, снижении познавательной способности следует провести диффдиагностику с идиопатическим синдромом Леннокса — Гасто, с миоклоническими формами детской эпилепсии.
Прогноз
Более благоприятный, чем при синдромах Уэста и Леннокса — Гасто, но не более чем в 50 % всех случаев.
Терапия
Первый выбор — Вальпроат, Этосуксимид, Ламотриджин. Начинать лечение следует с Вальпроата. Второй выбор — Бензодиазепины, Клобазам, Клоназепам. При присоединении ГТКП, особенно при резистентности к терапии — бромиды, применять АКТГ.
Респираторные аффективные судороги.
Эти пароксизмальные состояния чреваты ошибочной диагностикой. Дифференциация между респираторными аффективными приступами и эпилепсией строится на анамнестических данных и связи приступов с эмоциональными реакциями на фоне невротических проявлений. В отличие от эпилепсии, при этих приступах характерны провоцирующие факторы, крик перед судорогами, цианоз, появляющийся до судорог, опистотонус при нормальной ЭЭГ, хотя даже выявленная патология на ЭЭГ не решает вопроса дифференциации. АК при респираторных аффективных судорогах безрезультативны. Лечение должно быть направлено на устранение невротизирующих факторов.
Фебрильные судороги.
Этиология и патогенез
До сих пор нет общепринятого мнения о природе этих приступов. Существует мнение, что гиперпирексия провоцирует идиопатическую эпилепсию и Фебрильные судороги являются нередко результатом не столько экстрацеребральных, сколько церебральных процессов. Считается, что гиперпирексия является провоцирующим моментом в вызывании судорожного припадка на благоприятной для этого почве (перинатальная патология — до 50 %, инфекции, травмы — до 20 % и др.). Нередко в семьях обнаруживаются случаи аналогичных приступов.
Распространенность
Встречаются до 15 % в общей популяции. Среди 55 % детей, перенесших «беспричинные» детские судороги, у 27 %, перенесших фебрильные судороги, наблюдается эпилепсия.
Клиника
Приступы тонико-клонических судорог (всегда первично-генерализованных) строго связаны с возрастом, спонтанно прекращаются в 4–5 лет, развиваются только при высокой температуре. Продолжительность приступа не более двух минут. Наблюдаются у детей не более 4–5 раз, чаще только в исключительных случаях.
Затяжные фебрильные судороги могут стать причиной склероза аммонова рога с риском развития фокальной эпилепсии.
Диагностика
Основывается на типичности клиники, этиологии и данных ЭЭГ.
Дифференциальная диагностика
Дети нуждаются в тщательном исследовании и контроле. О возможности развития эпилепсии следует думать, если имеются указания в анамнезе на неврологические повреждения, фокальное начало приступов, и/или фокальности на ЭЭГ, а также при наличии более 4–5 приступов, при их появлении при температуре менее 38,5 град. С и при семейной предрасположенности к эпилепсии.
Прогноз
При первичных проявлениях в возрасте 3 лет рецидивы встречаются крайне редко. Почти у 30 % детей с фебрильными судорогами в дальнейшем развивается эпилепсия.
Терапия
При наличии доброкачественных форм нет необходимости в применении АК терапии. При затяжных фебрильных судорогах требуется целенаправленное применение АК и другое этиопатогенетическое лечение.
Синдром Леннокса — Гасто.
Этиология и патогенез
Относится (как и синдром Уэста) к мультифакторным эпилепсиям, то есть имеются подозрения на наличие симптоматической этиологии, но не подкрепляющиеся результатами морфологических исследований, и этиология в таком случае остается криптогенной. В Международной классификации эпилепсия выделена в разделе Генерализованные формы эпилепсии как криптогенная и симптоматическая. Нередко прослеживаются органические резидуальные церебральные синдромы (пре-, пери- и постнатальные), подострые энцефалопатии, нейрометаболические заболевания, туберозный склероз.
Распространенность
Проявляется у детей от 2 до 8 лет, но чаще в дошкольном возрасте, 2–6 лет.
Примерно 30 % таких приступов рекрутируются из случаев синдрома Уэста.
Клиника
Начало в возрасте от 2 до 8 лет, поздние формы от 10 до 20 лет.
Наиболее часты виды припадков — миоклонико-астатические припадки, атипичные абсансы, молниеносные кивательные судороги, внезапные падения, тонические приступы (обычно во сне). Нередко встречаются и генерализованные тонико-клонические, миоклонические, парциальные припадки. Имеется тенденция к серийности разнообразных приступов с состоянием ступора, а также незаметному переходу в эпилептический статус.
Неврология: в 40 % случаев — церебральные парезы и гипотонико-астатические нарушения.
Психика: обычно — умственная отсталость до степени тяжелой деменции, психоорганические нарушения. В 80 % случаев — тяжелые когнитивные и личностные нарушения органического типа.
Нейрорадиология и патоморфология: фокальные или диффузные структурные нарушения.
Диагностика
Основывается на типичной клинической картине и ЭЭГ данных. На ЭЭГ обычно имеются изменения фона в виде медленных пик-волн меньше 3 Гц, ночью серии пиков (доходит до 100 за ночь), часто — мультифокальные изменения. Ранее считалось, что для синдрома Леннокса — Гасто патогномонична картина ритмических комплексов «пик-волна» 2,5 Гц. На самом деле описание паттерна ЭЭГ при синдроме Леннокса — Гасто это та же гипсаримия, только с большим содержанием «острых» феноменов. Заключение по ЭЭГ о гипсаритмии подтверждает диагноз синдрома Леннокса — Гасто.
Дифференциальная диагностика
Синдром Уэста.
Прогноз
В 75 % случаев — резистентность к терапии. Возможно персистирование миоклонико-астатических припадков во взрослый возраст, переход в большие судорожные приступы. Неблагоприятные прогностические признаки — предшествующее органическое поражение мозга или синдром Уэста, распространенные и частые тонические судороги, наклонность к статусному течению.
Лечение
Обычно приступы купируются с трудом. Более чем в половине случаев синдром развивается на фоне предшествующей энцефалопатии, но в 40 % случаев возникает как бы первично.
Препараты первого выбора — Вальпроат, Этосуксимид. Второй выбор — Бензодиазепины, АКТГ, кортикостероиды. В последние годы препаратом первого выбора становятся Вигабатрин и Ламотриджин, которые избирательно увеличивают содержание в мозге тормозного нейротрансмиттера — ГАМК.
Салаамов тик.
В изолированном варианте — в виде ритмических движений головой в передне-заднем направлении, к которым присоединяются кивательные движения туловища в том же направлении и, иногда, с нистагмом. Эти движения медленные и возникают у лиц с интеллектуальной недостаточностью (олигофренов), особенно в сидячем положении, сериями по 20–30. В таком варианте эти состояния не имеют отношение к эпилепсии.
Салаамов тик следует отличать от салаамовых приступов, которыми обозначаются инфантильные (младенческие) спазмы или пропульсивные припадки при синдроме Уэста. При этом синдроме судороги в виде флексорных туловищных движений или даже более простых движений — «кивки», «клевки», «поклоны», «складывания» — по типу «перочинного ножа» (во франкоязычной литературе). Это как бы рудиментарные судороги, но именно судороги, насильственный поклон, а не падение головы вперед из-за утраты тонуса. Такая картина получается из-за незрелости механизмов кортико-спинального контроля.
ЭЭГ в более раннем возрасте — больше разрядов дельта-активности, а в более позднем возрасте, когда нейросинаптические медиаторные системы уже обеспечивают генерацию, — больше быстрых эпилептических феноменов.
Симптоматическая ранняя миоклоническая энцефалопатия (ранняя младенческая эпилептическая энцефалопатия с паттернами «вспышка-угнетение» [burst-suppression] на ЭЭГ, синдром Отахара).
Этиология
Заболевание относится к симптоматическим генерализованным эпилепсиям неспецифической этиологии. Часто встречаются семейные случаи заболевания, что указывает на какое-то нарушение метаболизма.
Распространенность
Синдром описан в 1976 году, определяется по очень раннему началу болезни, встречается редко.
Клиника
Начало в первые несколько месяцев жизни частым фрагментарным миоклонусом. Затем — парциальные припадки, массивные миоклонусы или тонические спазмы.
Диагностика
Основывается на клинических особенностях и ЭЭГ данных. На ЭЭГ — супрессивно-взрывчатая активность, которая может перейти в гипсаритмию.
Дифференциальная диагностика
Синдром Уэста.
Прогноз
Неблагоприятный. В возрасте 4–6 месяцев часто отмечается переход в синдром Уэста. Течение очень тяжелое и быстрое драматическое развитие. Психомоторное развитие прекращается и на первом же году может наступить смерть.
Терапия
Выраженная резистентность к лечению АК.
Синдром Уэста (эпилепсия с судорогами типа молниеносных «салаам»-поклонов, «инфантильные спазмы», пропульсивные припадки).
Этиология
Относится (как и синдром Леннокса — Гасто) к мультифакторным эпилепсиям. В Международной классификации эпилепсии выделена как криптогенная и симптоматическая в разделе «Генерализованные формы эпилепсии». Нередко прослеживаются органические резидуальные церебральные синдромы (пре-, пери- и постнатальные), подострые энцефалопатии, нейрометаболические заболевания, в 10 % случаев — туберозный склероз.
Синдром Уэста можно подразделить на 2 группы: симптоматическая группа — наличие предшествующих признаков поражения мозга (предшествующая приступам умственная отсталость, неврологические, нейрорадиологические изменения или другие типы припадков, или известная этиология) и идиопатическая (меньшая) группа.
Распространенность
Проявляется у детей возраста 3–7 месяцев жизни, чаще у мальчиков.
Клиника
Характерна триада признаков: инфантильные спазмы + задержка психомоторного развития + гипсаритмия. Один из признаков триады может выпадать.
Спазмы могут быть сгибательными, разгибательными, чаще же — смешанные. Приступы в большинстве случаев заключаются во внезапно начинающемся, напоминающим испуг, генерализованном миоклонусе с рывками вверх, поднятием головы, напоминающими реакцию Моро — молниеносные (длительность — до 1 с) клонические судороги со сгибанием головы и туловища, иногда — с падением на колени. В некоторых случаях приступы проявляются также в коротком, но серийном, кивательном движении головы («кивки»). Реже эти приступы протекают как при замедленной киносъемке, чем напоминают восточное приветствие «саалам». Всегда выявляется выраженная тенденция к серийности судорог, незаметному переходу к статусу, комбинирование с большими приступами.
Психика: задержка психомоторного развития.
Неврология: в 80 % случаев — церебральные парезы, гипотонико-атактические нарушения, микроцефалия.
Нейрорадиология: в 90 % случаев находят грубые структурные нарушения.
Патоморфология: микроцефалия, лиссэнцефалия, пахигирия, микрогирия, глиоматоз, глобарный склероз, сосудистые мальформации.
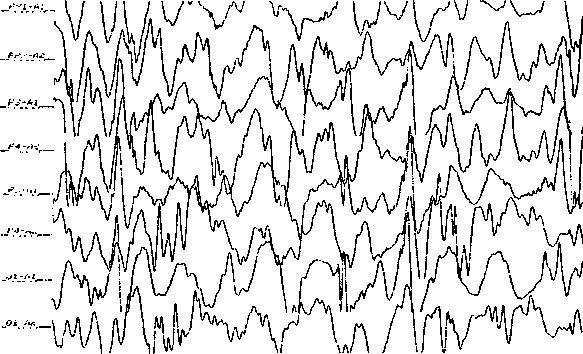
Гипсаритмия. Скорость — 30 мм/с. Амплитуда — уменьшена по сравнению с обычным усилением в 3 раза.
Диагностика
Основывается на типичной триаде клиники и патогномоничных ЭЭГ данных, активизирующихся во сне. ЭЭГ вне припадка — гипсаритмия. Гипсаритмия — это непрерывная генерализованная высокоамплитудная медленная и гиперсинхронная активность с острыми волнами, пиками, медленными пик-волновыми комплексами. ЭЭГ данным во время припадка при молниеносных миоклониях соответствуют генерализованные пики и острые волны, при тонических судорогах — низкоамплитудные высокочастотные генерализованные пики, нарастающие по амплитуде к концу припадка.
Дифференциальная диагностика
Синдром Леннокса — Гасто.
Прогноз
Чаще зависит от своевременного лечения АКТГ, но принципиально — от симптоматического или идиопатического характера синдрома. В основном прогноз неблагоприятный. Смертность отмечается примерно в 20 % случаев. В 90 % случаев — нарушение психического развития. Часто переход в синдром Леннокса — Гасто. Благоприятные прогностические признаки: нормальное психомоторное развитие к началу приступов, отсутствие других эпилептических проявлений, нормальный неврологический и нейрорадиологический статус, быстрый ответ на терапию и отсутствие рецидивов, отсутствие фокальных или мультифокальных проявлений на ЭЭГ после исчезновения гипсаритмии.
Большинство идиопатических случаев показывают благоприятный прогноз, если лечение начато своевременно.
Терапия
На первом этапе — высокие дозировки витамина В6, что проявляется уже в первые дни лечения. При неэффективности — Вигабатрин в высоких дозах. Если нет эффекта в течение 2 недель — начать прием Вальпроата, кортикостероидов. Побочные эффекты значительны (синдром Кушинга, нефролитиаз и др.).
Эпилепсия парциальная постоянная (Кожевникова) (G40.5).
Этиология
Кожевников описал 2 синдрома. Первый — собственно эпилепсия Кожевникова (epilepsia partialis continua) детского возраста, в этиологии которой лежит деструктивное локальное поражение мозга любой этиологии (опухоль, сосудистое, глиоз). Второй — хроническая прогредиентная Epilepsia Partialis continua (синдром Кожевникова детского возраста, синоним — синдром прогрессирующей энцефалопатии Расмуссена), где этиология вирусно-воспалительная.
При собственно эпилепсии Кожевникова (epilepsiapartialis conlinua) детского возраста
Клиника
Представлена парциальной непрогредиентной роландической эпилепсией у детей или у взрослых, связанной с повреждением моторной коры.
Возраст начала любой.
Приступы: фокальные моторные припадки, длящиеся в течение дней, недель, месяцев.
Психика: без особенностей.
Нейрорадиология: соответствующие этиологическому фактору морфологические изменения.
Неврология: клинические проявления соответствуют поражению коры и не имеют тенденции к прогрессу (резидуальная органика); прогрессирование указывает на опухоль.
Диагностика
Основывается на клинических проявлениях и ЭЭГ данных. На ЭЭГ — ограниченные эпилептиформные разряды в роландической области, контрлатеральной стороне судорожных проявлений.
Дифференциальная диагностика
Проводится на ранних этапах — с роландической эпилепсией, в дальнейшем — с локализованными симптоматическими формами.
Прогноз
Непрогредиентное течение, если не прогрессирует этиологический фактор.
Терапия
Первый выбор — Карбамазепин. Второй выбор — Вальпроат, бензодиазепины (клоназепам, клобазам).
Хроническая прогредиентная Epilepsia Partialis continua (синдром прогрессирующей энцефалопатии Расмуссена).
Клиника
Приступы имеют начало с фокальных моторных с последующим присоединением локальных миоклоний. Вначале приступы четко фокальны, затем локально непостоянны, тенденция к генерализации. Часто припадки во сне.
Возраст начала 2—10 лет.
Неврология: с развитием заболевания развивается прогредиентный гемипарез.
Психика: нарастание деменции, задержки психического развития.
Нейрорадиология: деструктивные изменения контрлатерально гемипарезу.
Диагностика
Основа — клиника и ЭЭГ данные. На ЭЭГ выявляются преимущественно диффузные дельта-волны с преобладанием в контлатеральном неврологическому проявлению полушарии, мультифокальные высокоамплитудные спайки, острые волны, пик-волны в больном полушарии, с последующим вовлечением второго.
Прогноз
Прогредиентное течение с развитием тяжелых неврологических и интеллектуальных дефектов.
Терапия
Первый выбор АК — Карбамазепин, второй выбор — Вальпроат, бензодиазепины. Медикаментозному лечению не поддается. При четко одностороней локализации требуется оперативное лечение (гемисферэктомия).
Первичная эпилепсия чтения (ЭЧ).
Этиология
В МКБ-10 не выделена. Редкая форма идиопатической фокальной эпилепсии с предположительной локализацией очага в теменно-височной области доминантного по речи полушария. Предполагается аутосомно-доминантное наследование при ЭЧ, и она имеет семейное накопление (до 40 %). Раньше относилась к рефлекторным фотосензитивным, однако тот факт, что приступы провоцируются даже во время чтения по системе Брайля, опровергло этот взгляд. В настоящее время считается, что пусковым механизмом приступа является трансформация графем в фонематическую речь.
Распространенность
ЭЧ — один из самых редких эпилептических синдромов. Частота встречаемости ЭЧ варьирует у народов, использующих разные системы письменности: максимальна для систем с буквенным написанием и минимальна для систем с иероглифическим. Отмечено преобладание больных мужского пола в соотношении примерно 2:1.
Клиника
Начало ЭЧ обычно приходится на пубертатный период и позже. Редко возникает у детей раннего школьного возраста.
Приступы возникают почти исключительно во время чтения, особенно вслух. Провокация связана с индивидуальными особенностями ситуации (содержание текста, характер артикуляции, освещенность). Наиболее частое проявление приступа — клонические подергивания в мышцах нижней челюсти, в жевательной мускулатуре, ощущения затруднения дыхания, «подавливания», или сенсорные нарушения, чаще в виде расплывающегося изображения. При продолжении чтения возможен переход в большой припадок.
Психика и неврология — без особенностей.
Диагностика
ЭЭГ в межприступном периоде в 80 % случаев регистрирует нормальную электроактивность. Фотосенситивность отмечена всего у 9 % больных, зато провокация пароксизмальной активности во время чтения наблюдается почти в 80 % случаев. Во время приступа обычно регистрируется билатерально-синхронная пик-волновая активность с амплитудным преобладанием в теменно-височных отделах доминантного полушария и/или генерализованные пик-волны.
Прогноз
В целом — благоприятный.
Терапия
Лечение АК оправдано, так как приступы имеют тенденцию со временем провоцироваться и другими факторами (разговор, игры, еда), даже могут стать спонтанными. Средства первого выбора — Вальпроат; второго выбора — Клоназепам. Имеются данные о хорошем эффекте блокатора кальциевых каналов — Флунаризина (Сибелиум) в качестве дополнительной терапии.
Эпилептический статус (Status epilepticus, SE) (G41).
Определяется как «стойкое эпилептическое состояние» с повторяющимися или непрерывными приступами, которые продолжаются более 30 минут или между которыми больной не может полностью достичь своего нормального психического и неврологического состояния.
Этиология
Этиологические факторы, определяющие развитие статуса, разнообразны. Статус может возникать как осложнение эпилепсии или быть ее манифестным проявлением. Основные причины возникновения эпилептического статуса без предшествующих эпилептических пароксизмов (de novo):
— нейроинфекции,
— острые нарушения мозгового кровообращения,
— черепно-мозговая травма,
— прогрессирующие заболевания ЦНС,
— интоксикации.
Распространенность
Эпилептический статус встречается с частотой 18–20 случаев на 100 000 населения и является одним из наиболее распространенных неотложных неврологических состояний. В 50 % случаев эпилептический статус возникает у детей раннего возраста. Среди больных эпилепсией статус также чаще отмечается у детей (10–25 %), чем у взрослых (5 %).
Классификация
Разновидности эпилептического статуса обозначаются в соответствии со встречающимися при нем формами приступов. Наиболее известны — статус судорожных припадков, статус малых припадков, статус сложных фокальных приступов, эпилепсия Кожевникова (G40.5), статус миоклонических приступов.
Прогноз
При SE представляет собой ситуацию, требующую неотложной помощи, так как связанная с ним смертность даже в настоящее время может доходить до 30–50 %.
Терапия
«Статус является истинным кризисом болезни (эпилепсии) и в меньшей степени вероятным ее завершением, которого нужно избегать с помощью правильного лечения…» (L.P. Clare, T.P. Prout, 1903).
Если ранее была диагностирована эпилепсия, то развитие SE всегда указывает на необходимость критического переосмысления стратегии применяемого медикаментозного лечения и прежде всего в тех случаях, когда не удается выяснить факторы, провоцирующие SE.
Эпилептический статус Grand mal (судорожных припадков) (Тонико-клонический эпилептический статус) (G41.0).
Этиология
Причины, чаще встречающиеся у взрослых (цереброваскулярные процессы, лишение алкоголя, гипоксические состояния), в детском возрасте почти не играют никакой роли. У детей этиологически доминируют менинго-энцефалические инфекции, врожденные аномалии развития, последствия церебральных повреждений, прогрессирующие нейродегенеративные заболевания. У новорожденных в подавляющем большинстве случаев статусы обусловлены нейрометаболическими нарушениями, инфекциями, кровоизлияниями в мозг, гипоксически-ишемическими энцефалопатиями, а в раннем грудном возрасте — острыми воспалениями и электролитными нарушениями.
У пациентов с выявленной ранее эпилепсией наиболее частой причиной статусов является снижение концентрации АК в крови (неправильная смена терапии, отмена антиконвульсантов). Наиболее частые «поставщики» статусов — лобнодолевые эпилепсии. Если статус встречается впервые, причиной может быть целый ряд основных заболеваний (опухоль мозга, энцефалит, церебро-васкулярная патология, черепно-мозговые травмы, интоксикации, алкоголизм, метаболические нарушения). Эти заболевания должны быть диагностированы и лечиться параллельно со статусом приступов.
Клиника
Частота судорожных приступов составляет от 3 до 20 в час. Основные критерии SE — наличие выраженных изменений, вызванных предшествующим припадком и относящихся к состоянию сознания, дыхания, гемодинамики. Сознание ко времени возникновения следующего припадка полностью не восстанавливается, и больной остается в состоянии оглушения, сопора или комы. При SE пролонгированном в клинике наступают изменения: длительность ГТКП уменьшается, коматозное состояние углубляется, судороги принимают тонический характер, гипотония мышц сменяется атонией, а гиперрефлексия — арефлексией. Нарастают гемодинамические и дыхательные нарушения. Наконец, судороги могут полностью прекратиться и наступает стадия эпилептической прострации: глазные щели и рот полуоткрыты, взор безучастный, зрачки широкие. В таком состоянии может наступить смерть.
Диагностика
SE изучен достаточно хорошо и его диагностика не вызывает затруднений при клиническом наблюдении.
Прогноз
Прогноз значительно зависит от его этиологии, так как смертность от статусов Grand mal в случаях ранее диагностированной эпилепсии — не 30–50 %, как при острых симптоматических, а только около 5 %. Вторым по важности прогностическим фактором является продолжительность статуса. Если статус длится более 30 минут, следует опасаться развития серьезных церебральных, сердечно-сосудистых, респираторных, вегетативных и метаболических осложнений (отек мозга, гипоксия, гипотензия, гиперпирексия, лактатацидоз, изменения электролитного баланса), которые приводят к необратимым неврологическим и нейропсихологическим нарушениям.
Терапия
В международной практике принято использовать унифицированную этапную схему со строго определенными временными рамками. На первом этапе применяется комбинированное лечение Диазепамом и Фенитоином, которое купирует статус больших приступов в 85–90 % случаев.
Этап 1 (0—10 мин.)
A) Необходимо обеспечить функции дыхания и кровообращения, при необходимости — кислородный зонд.
Б) Определить концентрацию противоэпилептического препарата в крови.
B) Измерить температуру.
Этап 2 (30–40 мин)
А) Диазепам 20 мг (детям 0,2–0,4 мг/кг/м.т.) ректально, или медленно внутривенно, или Клоназепам 2 мг (детям 0,01 — 0,04 мг/кг/ м.т.) медленно внутривенно. Следует учитывать быстрое наступление действия (5—15 мин), однако не только в плане противосудорожных эффектов, но и угнетения дыхания, седативного эффекта.
Б) Сразу после А) назначается внутривенно Фенитоин (детям — 10–15—20 мг/кг/м.т.), скорость инъекции — менее 50 мг/мин. Следует учитывать, что максимальный эффект наступит через 20–30 мин. При падении АД, возникновении аритмии скорость введения необходимо уменьшать. Часто первым симптомом интоксикации является нистагм.
В настоящее время на нашем рынке появляется инфузионная форма Фенитоина (например, Фенгидан фирмы «Деситин»).
Имеются «альтернативные» варианты международных стандартизированных схем, причем оговаривается, что «для лечения невосприимчивого к терапии статуса не существует надежных рекомендаций».
«Невосприимчивым», или резистентным (рефрактерным), считают статус, который продолжается 60 и более минут, несмотря на применение не менее двух антиконвульсантов первой очереди выбора. Эти варианты предполагают применение инфузионных форм Фенобарбитала или Лидокаина, или бензодиазепинов (Лоразепама, Паральдегида).
Фенобарбитал (детям: 4–6—10 мг/кг/м.т.) вводится внутривенно, скорость введения — менее 100 мг/мин. Следует учитывать возможность угнетения дыхания, седативный эффект, большой период полувыведения из организма.
Лидокаин — инъекция вводятся ударной дозы 100–200 мг внутривенно, затем — инфузия 3–4 мг/кг. Следует учитывать возможность аритмии, падения АД, реакции идиосинкразии, немедленное действие.
Лоразепам 4 мг (+4 мг через 10 минут) вводится внутривенно.
Следует учитывать возможность угнетения дыхания, седативный эффект, длительность действия около 12 часов.
Этап 3 (рефрактерный статус)
Применяется общая анестезия (наркоз), например с помощью тиопентала, который проводится в отделении интенсивной терапии (реанимации). Наркоз необходимо продолжать 12–24 часа после последнего приступа. Лучше, конечно, постоянно регистрировать ЭЭГ с целью подавления «вспышек».
Эпилептический статус Petit mal (Эпилептический статус абсансов, SEA) (G41.1).
Этиология
Эта форма статуса может быть первым проявлением эпилепсии, при котором у пожилых людей внезапно развивается спутанность сознания. Статус абсансов может последовать за «большим» приступом или перейти в него.
Распространенность
Наиболее часто встречается у детей. Может возникать в 5 % случаев генерализованных эпилепсии.
Клиника
SEA представляет собой разновидность генерализованного бессудорожного статуса. Состояние известно под устаревшими терминами: «статус спутанности», «пик-волновой ступор». Имеющиеся нарушения сознания выражены в различной степени — от легкого нарушения концентрации до дезориентации и ступора. В отдельных случаях изменение сознания столь незначительно, что может быть выявлено только при психологическом тестировании. Примерно у 50 % больных наблюдается дрожание век, подергивание рук и другие судорожные проявления. SEA в ряде случаев является причиной эпилептической фуги.
Различают две разновидности — статус типичных и статус атипичных абсансов — по ряду признаков, главным из которых являются ЭЭГ проявления.
Для статуса «типичных» абсансов клинически характерны:
— внезапное начало и окончание,
— продолжительность — до нескольких дней (обычно — меньше),
— трансоподобное состояние с отсутствием реакций на внешние стимулы,
— наличие в анамнезе идиопатических форм генерализованных эпилепсии.
Для статуса «атипичных» абсансов клинически характерны:
— постепенное начало и окончание, наличие продромального периода,
— продолжительность — до нескольких недель,
— сочетание атипичных абсансов с тоническими, миоклоническими приступами,
— наличие в анамнезе симптоматических или криптогенных генерализованных эпилепсии (чаще — синдрома Леннокса — Гасто).
Диагностика
Основывается на клинике и обязательном ЭЭГ исследовании. ЭЭГ всегда показывает комплексы пик-волн, более или менее непрерывные.
Частота разрядов и морфология комплексов может часто отличаться от их классического рисунка — 3 в с.
Для статуса «типичных» абсансов характерны на ЭЭГ — наличие генерализованных билатерально-синхронных комплексов «пик-волна», регулярно повторяющихся с частотой 3 в с.
Для статуса «атипичных» абсансов на ЭЭГ — наличие продолжительных медленных комплексов «пик-волн», нерегулярно повторяющихся с частотой 1,5–2 Гц.
Терапия
Вводятся в/в инъекции препаратов группы бензодиазепинов — Диазепам в дозе 10–20 мг, детям — 0,02 — 0,04 мг/кг/м.т., или ректальное введение 20–30 мг Диазепама. Грудным детям — 5 мг, детям с массой тела более 15 кг — 10–20 мг.
В последнее время применяют также в/в введение Вальпроатов. Терапия статуса абсансов должна проводиться по возможности при продолжающемся ЭЭГ контроле.






