Врожденные пороки развития плода
Врожденные пороки развития плода занимают 2-3-е место в структуре причин перинатальной гибели плода и новорожденного. Важное значение имеет ранняя диагностика пороков развития, которая необходима для своевременного решения вопроса о возможности продолжения беременности, что определяется видом порока развития, совместимостью с жизнью и прогнозом в отношении постнатального развития.
В зависимости от этиологии различают наследственные (генетические), экзогенные и мультифакториальные врожденные пороки развития плода.
К наследственным относят пороки развития, возникающие вследствие мутаций, т.е. стойких изменений наследственных структур в половых клетках (гаметах) или зиготе. В зависимости от того, на каком уровне произошла мутация (гены или хромосомы), выделяют моногенные синдромы и хромосомные болезни.
К экзогенным относят пороки, обусловленные повреждающим действием экзогенных факторов. Данные факторы, действуя в период гаметогенеза или беременности, приводят к возникновению врожденных пороков, не нарушая структуру наследственного аппарата.
Пороками мультифакториального происхождения называют пороки, возникшие под комбинированным воздействием генетических и экзогенных факторов.
Выделяют также изолированные (локализованные в одном органе), системные (в пределах одной системы органов) и множественные (в органах двух и более систем) пороки.
Пороки развития центральной нервной системы
Классификация наиболее часто встречающихся пороков развития ЦНС
1. Гидроцефалия:
• стеноз водопровода мозга
• открытая гидроцефалия
• синдром Денди-Уокера.
2. Папиллома сосудистого сплетения.
3. Дефекты нервной трубки:
• spina bifida;
• анэнцефалия;
• цефалоцеле.
4. Микроцефалия.
Гидроцефалия.
Гидроцефалия наблюдается с частотой 0,3-0,8 на 1000 живорожденных и проявляется обструкцией на одном из участков пути циркуляции цереброспинальной жидкости. Внечерепные аномалии при гидроцефалии встречаются в 63 %: агенезия и дисплазия почек, дефект межжелудочковой перегородки, тетрада Фалл о, менингомиелоцеле, расщепление верхней губы, твердого и мягкого неба, агенезия заднепроходного отверстия и прямой кишки, дисгенезия гонад. Гидроцефалия представлена в основном стенозом водопровода мозга (сужение сильвиева водопровода); открытой гидроцефалией (расширение желудочков мозга и субарахноидальной системы мозга в результате обструкции внежелудочковой системы путей оттока цереброспинальной жидкости); синдромом Денди-Уокера (сочетание гидроцефалии, кисты задней черепной ямки, дефектов червя мозжечка, через которые киста сообщается с полостью IV желудочка).
Папиллома сосудистого сплетения.
Это новообразование локализуется на уровне преддверия боковых желудочков. Папиллома сосудистого сплетения представлена тканью ворсин, гистологически сходной с тканью интактного сосудистого сплетения, и имеет доброкачественное течение. Как правило, она сочетается с гидроцефалией. Папиллому сосудистого сплетения диагностируют с помощью нейросонографии или компьютерной томографии.
Дефекты нервной трубки.
Данный термин объединяет анэнцефалию, цефалоцеле и spina bifida.
Spina bifida – срединный дефект дорсальных дуг позвонков, сопровождающийся обнажением содержимого спинномозгового канала. Spina bifida встречается у 4,1 из 1000 родившихся. Эта аномалия наследуется по многофакторному типу. Spina bifida может быть частью генетических синдромов (с изолированным мутантным геном) или хромосомных аномалий (трисомии по 13-й и 18-й парам хромосом, триплоидия, несбалансированная транслокация либо кольцевая хромосома), результатом воздействия на плод тератогенных факторов в период органогенеза. Различают spina bifida cystica (кистозная форма спинномозговой грыжи с образованием грыжевого мешка, содержащего оболочки мозга и/или вещество мозга) и spina bifida occulta (скрытая форма, которая не сопровождается образованием грыжевого выпячивания). Спинномозговая грыжа часто сочетается с гидроцефалией, врожденными пороками сердца и мочеполовой системы.
Прогноз зависит от уровня и степени поражения, наличия сопутствующих аномалий. Выживаемость детей, получивших лечение в раннем неонатальном периоде, не более 4 0%, причем 25% из них остаются парализованными. При выявлении патологии и наличии нежизнеспособного плода показано прерывание беременности. Показанием к досрочному прерыванию беременности является быстрое нарастание вентрикуломегалии и макрокрании.
Анэнцефалия
Анэнцефалия (псевдоцефалия, экстракраниальная дисэнцефалия) – отсутствие полушарий мозга и большей части свода черепа, при этом наблюдается дефект лобной кости выше супраорбитальной области, височная и часть затылочной кости отсутствуют. Верхняя часть головы покрыта сосудистой мембраной. Структуры среднего и промежуточного мозга частично или полностью разрушены. Гипофиз и ромбовидная ямка в основном сохранены. К типичным проявлениям можно отнести выпученные глаза, боль-шой язык и короткую шею. Данная патология встречается с частотой 3,6 на 1000 родившихся. Чаще ее обнаруживают у новорожденных девочек. Отмечаются многофакторное и аутосомно-рецессивное наследование, хромосомные аномалии. К факторам риска относят сахарный диабет у матери. В экспериментах на животных установлена тератогенность радиации, салицилауов, сульфаниламидов, повышенного содержания углекислого газа, Лхографический диагноз может быть установлен уже в 12- нед беременности. Среди родившихся 32 % являются живорожденными. При внутриутробной диагностике анэнцефалии прерывание беремен-ности показано при любом сроке.
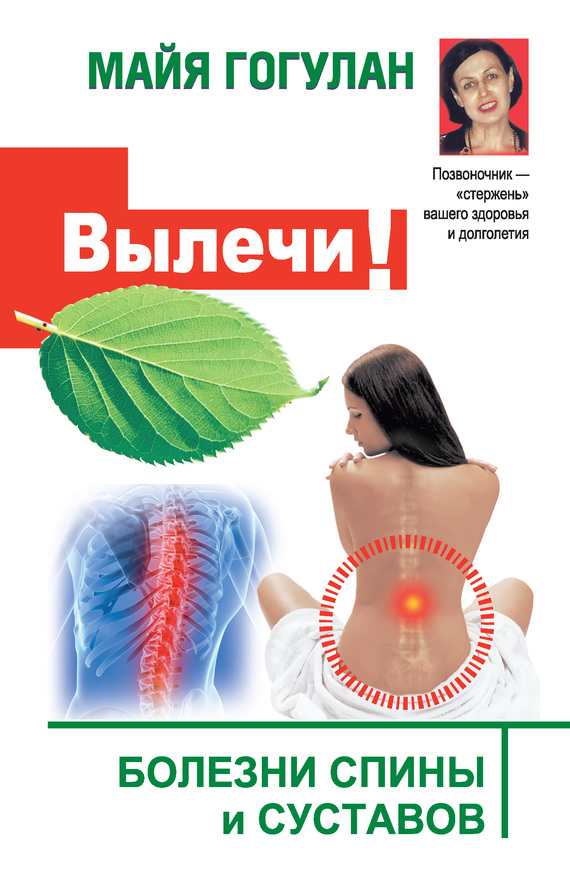
Цефалоцеле (энцефалоцеле, краниальное или окципитальное менингоПеле, расщепление черепа) – выбухание содержимого черепной коробки Через костный дефект (рис.16.1). Термином "краниальное менингоцеле" обозначают выпячивание через дефект только менингеальных оболочек. При. нахождении в грыжевом мешке ткани мозга применяют термин "энцефалоцеле". Цефалоцеле встречается редко и является компонентом многих генетических (синдромы Меккеле, срединного расщепления лица) и негенетических (амниотические перетяжки) синдромов. Прогноз зависит от наличия ткани мозга в грыжевом мешке и сопутствующих гидроили микроцефалии. При энцефалоцеле летальность достигает 44 %, при менингоцеле – не наблюдается. Нормальное интеллектуальное развитие установлено только у 9 % детей с энцефалоцеле и у 60 % – с менингоцеле. Показано прерывание беременности при любом сроке.
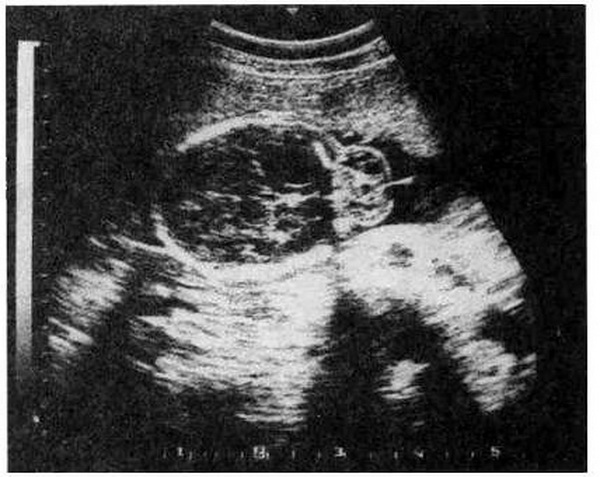
Рис. 16.1. Энцефалоцеле, диагностированное с помощью УЗИ при сроке беременности 27 нед.
1 – костный дефект; 2 – энцефалоцеле (поперечное сечение головки плода).
Микроцефалия (микроэнцефалия).
Микроцефалия – клинический с и т дром, для которого характерны уменьшение окружности головки и умственная отсталость; встречается с частотой 1,6 на 1000 живорожденных. Микроцефалия является полиэтиологическим заболеванием, в развитии которого важную роль играют генетические (хромосомные аберрации, моногенные дефекты) и экологические факторы. Прогноз зависит от наличия сочетанных аномалий. Трисомии по 13-й, 18-й хромосоме, синдром Меккеля относятся к фатальным поражениям. В отсутствие сопутствующих аномалий прогноз зависит от размеров головки: чем она меньше, тем ниже индекс интеллектуального развития. Микроцефалия относится к неизлечимым заболеваниям. Акушерская тактика – прерывание беременности.
Пороки развития почек и мочевыводящих путей
Поликистозная болезнь почек инфантильного типа (поликистозная болезнь почек I типа, аутосомно-рецессивное поликистозное заболевание почек) проявляется двусторонним симметричным увеличением почек в результате замещения паренхимы вторично расширенными собирательными канальцами без пролиферации соединительной ткани. Прогноз неблагоприятный. Смерть наступает от почечной недостаточности. Акушерская тактика заключается в прерывании беременности при любом сроке.
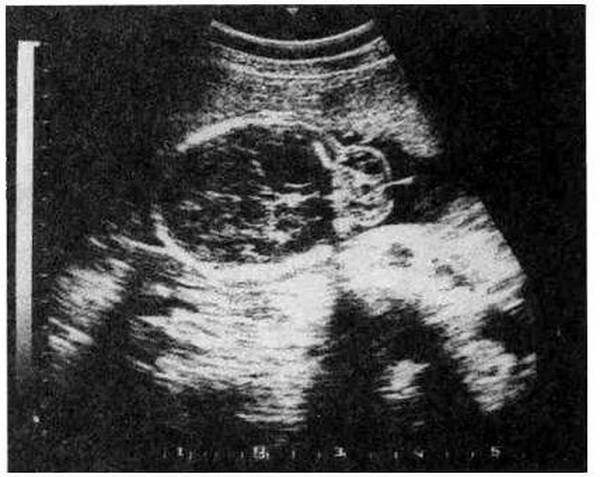
Рис. 16.2. Поликистоз правой почки плода (срок беременности 27 нед).
Поликистозная болезнь почек взрослого типа (аутосомно-доминантная болезнь, гепаторенальная поликистозная болезнь взрослого типа, синдром Поттера III типа) характеризуется замещением паренхимы почки многочисленными кистами разных размеров, которые образуются вследствие расширения собирательных канальцев и других канальцевых сегментов нефрона. Почки поражены с двух сторон и увеличены, но односторонний процесс может быть первым проявлением заболевания. Печень также вовлекается в патологический процесс – развивается перипортальный фиброз, который имеет очаговый характер.
Этиология заболевания неизвестна. Однако тип наследования обусловливает 50 % риск развития болезни, а ее генетический фокус расположен в 16-й паре хромосом. Мутантный ген носит один из 1000 человек. Пенетрация гена происходит в 100 % случаев, однако течение заболевания может варьировать от тяжелых форм со смертельным исходом в неонатальном периоде до бессимптомных, обнаруживаемых только на вскрытии.
Поликистоз почек хроническое заболевание, первые симптомы его появляются в возрасте 35 лет (постоянные боли в поясничной области, увеличение почек, артериальная гипертензия, почечная недостаточность и уремия). Акушерская тактика заключается в ранней диагностике и прерывании беременности. Пренатальную диагностику осуществляют путем биопсии ворсин хориона.
Поликистоз почек (мультикистозная болезнь, кистозное заболевание почек, синдром Поттера II типа, диспластическая болезнь почек) характеризуется кистозным перерождением почечной паренхимы вследствие первичного расширения почечных канальцев (рис. 16.2). Процесс может быть двусторонним, односторонним и сегментарным. Заболевание возникает в основном спорадически и может быть вторичным в комплексе с другими синдромами. Акушерская тактика при двустороннем процессе, диагности-рованном в ранние сроки, в связи с неблагоприятным прогнозом заключается в прерывании беременности. При одностороннем процессе и нормальном кариотипе без сочетанных аномалий показано обычное родоразрешение с последующей консультацией ребенка у специалиста.
Врожденный гидронефроз (пиелоэктазия) является следствием обструкции мочевыводящих путей в месте соединения почечной лоханки и мочеточника. Частота его развития не установлена, так как эта патология представляет собой спорадический феномен. После рождения у мальчиков его диагностируют в 5 раз чаще. У 27 % детей с гидронефрозом выяапяют пузырно-мочеточниковый рефлюкс, двустороннее удвоение мочеточников, двусторонний обструктивный мегауретер, нефункционирующую контралатеральную почку и ее агенезию, у 19 % – аномалии развития различных органов. Акушерская тактика зависит от времени возникновения и длительности течения патологического процесса, а также степени нарушения функции почек.
Врожденные пороки сердца
Частота врожденных пороков сердца (ВПС) составляет от 1-2 до 8-9 на 1000 живорожденных. Наиболее распространенными из ВПС являются дефекты межпредсердной и межжелудочковой перегородок, открытый артериальный проток, стеноз легочной артерии, гипопластический синдром левых отделов сердца, единственный желудочек и др. В 90 % случаев ВПС являются результатом многофакторного повреждения (генетическая предрасположенность и средовые факторы). Риск повторения порока составляет 2-5 % после рождения одного и 10-15 % – двух больных детей. Моногенное наследование отмечается у 1-2% детей с ВПС. У 5% детей обнаруживают хромосомные аномалии, из которых основными являются трисомии. У 1-2 % новорожденных отмечено влияние различных тератогенов.
Эхо кардиографическое исследование плода представляет собой наиболее информативный метод пренатальной диагностики ВПС. Показания к пренатальной диагностике определяются состоянием матери и плода.
1. Показания, обусловленные состоянием матери:
• наличие ВПС у членов семьи
• сахарный диабет
• прием беременной лекарственных препаратов во время органогенеза
• алкоголизм
• системная красная волчанка
• фенилкетонурия.
2. Показания, обусловленные состоянием плода:
• многоводие
• неиммунная водянка
• дизаритмии
• экстракард и альные пороки;
• хромосомные нарушения.
• симметричная форма внутриутробной задержки роста плода.
Прогноз зависит от вила порока, наличия сопутствующих аномалий и хромосомных нарушений.
Акушерская тактика заключается в том, что после тщательного эхокардиографического исследования проводят кордоили амниоцентез с целью получения материала для хромосомного анализа. В случае выявления ВПС у нежизнеспособного плода показано прерывание беременности. При доношенной беременности лучше проводить родоразрешение в специализированных перинатальных центрах. При сочетанных пороках и генетических аномалиях необходимо прерывание беременности при любом сроке.
Аномалии формирования стенок брюшной полости и пороки развития желудочно-кишечного тракта
Диафрагмальная грыжа представляет собой перемещение органов брюшной полости в грудную полость через дефект диафрагмы.
Предполагают, что врожденная диафрагмальная грыжа – следствие тератогенного воздействия хинина, противоэпилептических препаратов, недостатка витамина А.
Врожденные диафрагмальные грыжи часто сочетаются с анэнцефалией, цефалоцеле, spina bifida, расщеплением губы и верхнего неба, омфалоцеле, дефектами межжелудочковой перегородки и тетрадой Фалло. Диафрагмальные грыжи всех типов, за исключением релаксации диафрагмы (заднелатеральной и позадигрудинной), могут сочетаться с хромосомными аномалиями (чаще трисомиями).
Прогноз неблагоприятный. Мертворождение отмечается в 35 % случаев, причем у 90 % мертворожденных выявляются несовместимые с жизнью сочетанные аномалии ЦНС и сердечно-сосудистой системы. Значительное число детей умирают в первые часы после рождения, из них 35 % в течение 1-го часа.
Акушерская тактика заключается в прерывании беременности до достижения плодом жизнеспособности. В случае родоразрешения в срок при доношенной беременности следует предусмотреть необходимость хирургической помощи ребенку.
Омфалоцеле (пупочная грыжа) – дефект передней брюшной стенки в области пупочного кольца, при котором образуется грыжевой мешок с енутрибрюшным содержимым, покрытый амниоперитонеальной мембраной.
Частота омфалоцеле составляет 1 на 5130-5800 родов. Данной патологии в 35-58 % случаев сопутствуют трисомии, в 47 % – врожденные пороки сердца, в 40 % – пороки развития мочеполовой системы, в 39 % – дефекты нервной трубки. Задержку внутриутробного роста плода выявляют в 20 % случаев.
Прогноз зависит от наличия сопутствующих аномалий. Перинатальные Потери чаще связаны с ВПС, хромосомными аберрациями и недоношенностью. Небольшой дефект устраняют путем одноэтапной операции, при большом производят многоэтапные операции с целью закрытия отверстия в передней брюшной стенке силиконовой или тефлоновой мембраной.
Акушерская тактика определяется сроком выявления порока, наличием °четанных аномалий и хромосомных нарушений. При обнаружении порока в ранние сроки беременности ее следует прервать. В случае выявления сопутствующих аномалий, несовместимых с жизнью, необходимо прерывать беременность при любом сроке. Метод родоразрешения зависит от жизнеспособности плода, так как в процессе родов при больших омфалоцеле могут произойти разрыв грыжевого мешка и инфицирование внутренних органов плода.
Гастрошизис – дефект передней брюшной стенки в околопупочной области с эвентрацией петель кишок, покрытых воспалительным экссудатом Частота гастрошизиса составляет 1:10 000 -1:15 ООО живорожденных. Аномалия встречается спорадически, однако отмечаются случаи семейного заболевания с аутосомно-доминантным типом наследования.
Сочетанные аномалии встречаются редко, но у 25 % детей наблюдаются вторичные изменения в ЖКТ, являющиеся следствием сосудистых нарушений, – незавершенный поворот кишечника, атрезия или стеноз его отделов. В 77 % случаев гастрошизису сопутствует задержка роста плода.
Летальность новорожденных составляет 8-28 %, а при расположении печени за пределами брюшной полости достигает 50 %.
При выявлении гастрошизиса до наступления периода жизнеспособности плода следует произвести прерывание беременности. При доношенной беременности роды проводят в учреждении, где может быть оказана хирургическая помощь.
Атрезия пищевода (с трахеопищеводным свищом и без него) – отсутствие сегмента пищевода, которое сопровождается образованием фистулы между ним и дыхательными путями. Этиология неизвестна. Частота патологии варьирует от 1:800 до 1:5000 живорожденных.
В 58 % случаев атрезии пищевода сопутствуют ВПС, генетические аномалии, пороки развития мочеполовой системы и другие аномалии ЖКТ.
Антенатальная диагностика затруднена, так как желудок содержит секрет желез или наполняется через фистулу. Вероятность установления правильного диагноза составляет 10 %. Прогноз зависит от наличия сопутствующих аномалий и респираторных осложнений, массы тела и гестационного возраста при рождении.
Для определения акушерской тактики проводят тщательную ультразвуковую оценку анатомии и топографии внутренних органов плода, включая эхокардиографию. При нежизнеспособном плоде показано прерывание беременности. Выявление сопутствующих несовместимых с жизнью аномалий является показанием к прерыванию беременности при любом сроке. При изолированной атрезии пищевода родоразрешение проводят через естест-венные родовые пути.
Атрезия двенадцатиперстной кишки – наиболее частая причина непроходимости тонкой кишки. Частота аномалии составляет 1:10.000 живорожденных. Этиология неизвестна. Возможно возникновение порока под воздействием тератогенных факторов. Описаны семейные случаи пилородуоденальной атрезии с аутосомно-рецессивным типом наследования. У 30-52 % больных аномалия изолированная, а в 37 % случаев обнаруживают пороки развития костной системы: аномальное число ребер, агенезия крестца, конская стопа, VI поясничный позвонок, двустороннее отсутствие первых цев кистей, двусторонние шейные ребра, слияние шейных позвонков. В 2 тЬ случаев диагностируют сочетанные аномалии ЖКТ: незавершенный поворот желудка, атрезия пищевода, подвздошной кишки и заднепроходного отверстия, транспозиция печени. У 8-20 % больных выявляют ВПС, приблизительно в '/з случаев атрезия двенадцатиперстной кишки сочетается с трисомией по 21-й паре хромосом.
Для определения акушерской тактики проводят детальную ультразвуковую оценку анатомии внутренних органов плода и его кариотипирование. До наступления периода жизнеспособности плода показано прерывание беременности. При обнаружении изолированной аномалии в Ш триместре возможно пролонгирование беременности с последующим родоразрешением и хирургической коррекцией порока развития.
Аномалии лицевых структур, шеи и костной системы плода
Расщелина лица (расщепление верхней губы и неба) представляет собой линейный дефект, распространяющийся от края губы до носового отверстия. Расщелина неба, сочетающаяся с расщелиной губы, через альвеолярные отростки и твердое небо может распространиться на носовую полость или даже на дно глазницы. Двусторонняя расщелина губы наблюдается в 20 % случаев, расщелина губы и неба – в 25 %. Расщелина лица составляет около 13 % от всех пороков развития и регистрируется с частотой 1:700 новорожденных.
Сочетанные аномалии обнаруживают в 50 % случаев при изолированной расщелине неба и только в 13 % – при расщелине губы и неба. Пренатальное выявление дефекта при помощи эхографии затруднено, однако благодаря проведению ультразвукового сканирования и цветного картирования возможности его диагностики расширяются. В отдельных случаях возможна диагностика аномалии с помощью фетоскопии. В отсутствие сочетанных аномалий используется общепринятая акушерская тактика независимо от срока диагностики.
Расщепление верхней губы (заячья губа) не препятствует акту сосания и представляет собой только косметический дефект. При сочетании расщепления верхней губы, челюсти и твердого неба (волчья пасть) отмечаются функциональные нарушения: при сосании молоко вытекает через нос вследствие сообщения его с полостью рта; молоко может попадать в дыхательные пути. Прогноз благоприятный: современные хирургические методы позволяют добиться коррекции косметических и функциональных дефектов.
Срединная расщелина губы (полная срединная расщелина губы, псевдомедиальная расщелина губы, премаксиллярная агенезия) – четырехугольный или треугольный дефект верхней губы, встречается в 0,2-0,7 % всех случаев расщелины губы. Аномалия встречается только как компонент двух синдромов: орбитального гипотелоризма (голопрозэнцефалия) и орбитального гипертелоризма. При эхографии проводят оценку внутричерепных структур, так как имеется связь между развитием структур лица и процессами дифференциации переднего мозга. Прогноз определяется только сочетанием с другими аномалиями. Интеллект у 80 % больных не нарушен.
Кистозная гигрома (лимфангиома или последствие обструкции яремного лимфатического ствола) представляет собой осумкованное скопление жидкости. Характеризуется наличием единичных или множественных кист мягких тканей в области шеи, образующихся вследствие нарушений в лимфатической системе. Кистозные гигромы встречаются в 1 случае на 200 спонтанных выкидышей (копчико-теменной размер плода более 30 мм). Кистозная гигрома часто сочетается с хромосомными аберрациями {синдром Тернера, трисомии по 13-й, 18-й, 21-й паре хромосом, мозаицизм). Как изолированная аномалия наследуется по аутосомно-рецессивному типу.
Прогноз: в большинстве случаев плод погибает в первые два триместра беременности. Около 90 % новорожденных нуждаются в хирургическом лечении, у 31 % развиваются нарушение глотания и обструкция дыхательных путей. Парез лицевого нерва вследствие хирургического лечения возникает у 24 % пациентов.
Акушерская тактика заключается в прерывании беременности при ранней диагностике патологии, при доношенной беременности роды ведут через естественные родовые пути.
Пороки развития костной системы.
Среди врожденных пороков костной системы наиболее часто встречаются амелия (аплазия всех конечностей) фокомелия (недоразвитие проксимальных отделов конечностей, при этом кисти и стопы соединяются непосредственно с туловищем), аплазия одной из костей голени или предплечья, полидактилия (увеличение числа пальцев на конечности), синдактилия (уменьшение числа пальцев вследствие сращения мягких тканей или костной ткани рядом расположенных пальцев), аномальная установка стоп, остеохопдродисплазии, характеризующиеся ано-малиями роста и развития хрящей и/или костей (ахондрогенез, ахондроплазия, танатоформная дисплазия, несовершенный остеогенез, гипофосфатазия и др.).
Наиболее важна диагностика пороков, несовместимых с жизнью. Многие формы скелетных дисплазий сочетаются с гипоплазией легких, обусловленной небольшими размерами грудной клетки вследствие недоразвития ребер. Развитие легочной недостаточности при этом может быть причиной смерти детей в первые часы внеутробной жизни.