Отчего и почему?
Большое количество вирусов населяет нашу планету. У разнообразных хозяев формируют они латентную инфекцию, и механизмы, поддерживающие вирусную персистенцию, тоже могут быть различными. В 1950 году американский вирусолог Р. Магнус изучал размножение вируса гриппа в курином эмбрионе. Ученый обратил внимание на то, что при заражении эмбрионов очень большой дозой вируса в них накапливается значительное количество вирусных частиц, при этом их инфекционность резко падает. Магнус объяснил обнаруженное несоответствие накоплением в эмбрионе «неполного» вируса, т. е. вирусных частиц, внутри которых отсутствует нуклеиновая кислота. Однако оказалось, что это не совсем так. Позднее выяснилось, что при условии массированного заражения куриных эмбрионов вирусом гриппа накапливается «урожай» потомства, среди которого преобладают вирусные частицы с нехваткой (дефектом) генетического материала. Иными словами, в разных частицах недостает того или иного кусочка (гена) рибонуклеиновой кислоты, которая служит генетическим материалом частиц вируса гриппа.
Такие вирусные частицы, не способные к размножению, получили название дефектных. Их поведение напоминает таковое «собаки на сене»: они не размножаются сами и, находясь внутри клетки, препятствуют размножению стандартных вирусных частиц. Однако при одновременном заражении клетки как дефектной, так и стандартной частицами картина меняется: дефектные частицы начинают размножаться, используя недостающие «инструменты» стандартных частиц. Поэтому роль дефектных вирусных частиц в механизме формирования персистенции можно представить следующим образом (рис. 5).
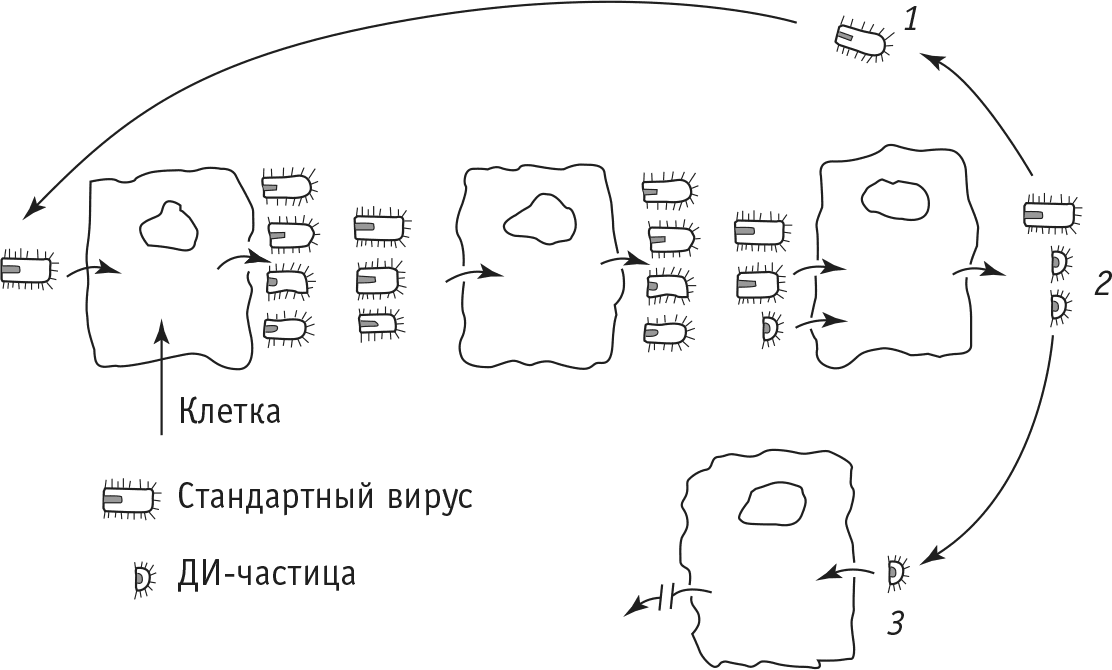
Рис. 5. Схема образования в клетках дефектных интерферирующих (ДИ) вирусных частиц: 1 – стандартная частица заражает клетку, которая после этого дает новый «урожай» стандартных частиц; 2 – дефектная частица заражает клетку совместно со стандартными частицами, в результате чего в клетке образуется «урожай» новых вирусных частиц, среди которых дефектные будут преобладающими; 3 – клетка, зараженная только дефектными частицами, не будет давать «урожая» вирусного потомства, и в таких клетках не будет происходить размножение стандартных частиц
Процесс последовательных заражений клеток (и в клеточной культуре, и в организме) обладает одной особенностью. Очень редко случается так, чтобы в клетке, зараженной стандартной частицей, среди накапливающегося потомства вируса появлялась хотя бы одна дефектная частица. Но если это все же происходит, то дефектные частицы, используя недостающие «инструменты» стандартных частиц, имеющиеся тут же, в клетке («под рукой»), начинают активно размножаться и делают это значительно быстрее, чем стандартные частицы, так как у дефектных генетического материала меньше, а стало быть, и собрать его можно быстрее. Выйдя из клетки вместе со стандартными частицами, дефектные теперь уже заражают новые клетки, в которых стандартные частицы уже размножаться не смогут, так как места в клетке уже «заняты». Такое явление называют интерференцией или исключением активности. Вместе с тем при одномоментном заражении новых клеток и дефектными, и стандартными частицами начинается одновременное размножение как тех, так и других, и опять дефектные будут опережать в этом стандартные вирусные частицы. Понятно, что очень скоро среди всего потомства вируса дефектные частицы станут преобладающими и будут защищать от стандартного вируса те клетки, в которых они (дефектные) находятся.
При определенном соотношении скорости процессов активного синтеза дефектных вирусных частиц и синтеза весьма небольшого количества стандартных частиц инфекционный процесс начинает протекать в латентной форме.
Подтверждением того, что описанное выше совершается именно так, а не иначе, служат следующие факты. Во-первых, дефектные интерферирующие вирусные частицы обнаружены в популяциях очень многих вирусов – гриппа человека, полиомиелита, бешенства, везикулярного стоматита, лимфоцитарного хориоменингита, клещевого энцефалита, японского энцефалита, лихорадки долины Рифт, гриппа птиц, парагриппа, обезьяньих вирусов, аденовирусов и многих других. Во-вторых, при ряде скрытых вирусных инфекций удалось выделить дефектные интерферирующие частицы из организма зараженных животных и человека, а также из зараженных клеточных культур.
В 1977 году в Москве в Институте эпидемиологии и микробиологии им. ?. Ф. Гамалеи Л. А. Денисов привел прямые доказательства ведущей роли дефектных частиц вируса гриппа в формировании и поддержании латентной формы инфекции. Ученый разработал метод обогащения вирусного препарата (по выбору исследователя) дефектными или стандартными вирусными частицами. Используя свой метод, он заражал как теми, так и другими препаратами вируса клеточные культуры, которые до этого считались нечувствительными к вирусу гриппа. И вот что было обнаружено.
При заражении клеток большой дозой вируса, обогащенного стандартными частицами, в так называемых нечувствительных клетках развивалась острая инфекция – клетки дегенерировали и погибали, а в питательную среду выходило большое количество частиц вновь синтезированного вируса гриппа. Если же такие клетки заражались тем же самым препаратом вируса, но в маленькой дозе, то формировалась скрытая инфекция, при которой клетки продолжали хорошо размножаться, не обнаруживали ни малейших признаков заболевания, но вместе с тем из таких клеток в питательную среду постоянно выделялся инфекционный вирус гриппа.
Но вот исследователь заразил клетки препаратом вируса, обогащенным дефектными частицами – в клетках не было обнаружено ни признаков заболевания, ни признаков присутствия вируса. Тогда Л. А. Денисов заражает клетки таким же препаратом, но в очень большой дозе. И вновь та же картина. Но что интересно: после заражения клетки стали размножаться быстрее и оказались устойчивыми даже к заражению стандартными частицами вируса. Прошло немало дней напряженного поиска, прежде чем Л. А. Денисову в содружестве со специалистом по электронной микроскопии из Института вирусологии им. Д. И. Ивановского А. А. Маныкиным удалось обнаружить в клетках такой культуры рибонуклеопротеидные (рибонуклеиновая кислота, связанная с белком) структуры вируса гриппа, которые, однако, были значительно короче, чем подобные структуры внутри стандартных вирусных частиц. Таким образом, латентная гриппозная инфекция этих клеток была доказана!
Проведенные эксперименты показали: изменяя в одном и том же препарате соотношение дефектных и стандартных вирусных частиц, можно получать различные формы инфекционного процесса, а присутствие в таком препарате большого количества дефектных частиц вызывает формирование латентной инфекции.
У вдумчивого читателя вполне естественно возникнут следующие вопросы: для чего дефектные интерферирующие вирусные частицы созданы природой? Почему в одной и той же вирусной «семье» существуют «два брата», из которых один (инфекционные частицы) «помогает» другому, а этот другой (дефектные частицы) только и делает, что «платит за помощь» постоянным стремлением помешать?
Пытаясь ответить на эти вопросы, мы должны будем коснуться столь важной проблемы, как саморегуляция у вирусов. Представьте ситуацию, при которой дефектных частиц нарабатывается все больше и больше. К чему это приведет? Совершенно ясно – к постепенному уменьшению количества стандартного вируса, так как мы помним, что в клетку, занятую дефектной частицей, стандартная частица попасть не может. Но подобный ход развития событий тоже не может остаться без последствий: стандартного вируса будет нарабатываться все меньше и меньше, все большее число клеток будет заниматься дефектными вирусными частицами, но сами-то они не способны к размножению… Им требуется помощник в лице стандартной вирусной частицы, а помощников стало очень мало. Поэтому интенсивность размножения дефектных частиц будет быстро снижаться. А следовательно, меньше клеток будет заражаться дефектными частицами, и в свободные от них клетки с успехом проникнут стандартные вирусные частицы. В связи с этим продукция стандартных вирусных частиц вновь увеличится. И до каких пор это будет происходить? До тех пор, пока среди стандартных частиц не появятся дефектные, а они появляются тем чаще, чем более массивной дозой заражается клетка. Появившиеся дефектные частицы начинают успешно размножаться, так как в этот момент у них много помощников (т. е. стандартных частиц), число дефектных частиц вновь возрастает, и все повторяется сначала. Таким образом, в самой популяции вируса заложен механизм, регулирующий и численность популяции, и ее качество.
Однако объяснить механизм формирования и длительного поддержания персистенции вирусов в организме хозяина присутствием и своеобразным эффектом дефектных вирусных частиц можно не всегда.
Уже шла речь о том, что много лет назад была открыта латентная вирусная инфекция у бактерий – лизогения, при которой дезоксирибонуклеиновая кислота (генетический материал) бактериофага объединяется (интегрирует) с хромосомой бактериальной клетки и в дальнейшем расщепляется как составная часть хромосомы в процессе клеточного деления. Наличие строгих доказательств интеграции генетического материала бактериофага с генетическим материалом бактериальной клетки и широкая распространенность лизогенных бактерий в природе наводили на мысль о возможности существования подобных взаимоотношений между вирусами животных и клетками животного организма.

Лев Александрович Зильбер
В 1946 году отечественный вирусолог Л. А. Зильбер (см. фото) выдвинул вирусную теорию происхождения опухолей, согласно которой вирус играет роль пускового механизма злокачественного процесса. Позднее в результате больших успехов в изучении лизогении Л. А. Зильбер значительно расширил и уточнил положения теории, названной им вирусо-генетической теорией происхождения опухолей. Ее главным постулатом как раз и стал факт объединения генетического материала опухолевого вируса с генетическим материалом клетки человека или животных.
Прошло много лет. В 1968 году в лаборатории американского вирусолога Р. Дюльбекко было доказано, что два известных опухолевых вируса – вирус полиомы и обезьяний 0В40 – поддерживают скрытую форму инфекции в клетках благодаря интеграции их ДНК с ДНК животной клетки. Публикации этих работ положили конец многолетним спорам о возможности существования для вирусов животных такого же типа взаимодействия с клеткой, как хорошо и давно известный в области бактериофагии. А спустя несколько лет интеграционный механизм был выявлен также и при латентной инфекции клеточных культур вирусами герпеса и аденовирусами.
Казалось бы, наступила пора торжества положений вирусо-генетической теории происхождения опухолей. Однако все эти открытия были связаны с вирусами, генетический материал (геном) которых представлен ДНК. Так как геном клетки высших организмов также представляет собой молекулу ДНК, то возможность интеграции двух геномов одинаковой химической природы не вызывала сомнений. И вот тут-то мы подходим, казалось бы, к наиболее уязвимому месту вирусо-генетической теории происхождения опухолей. Ведь большинство скрытых опухолевых вирусов животных являются РНК-содержащими! Это было давно известно и, откровенно говоря, очень затрудняло теоретическое объяснение возможной интеграции геномов таких вирусов с клеточным геномом.
Работы по разрешению этого кажущегося противоречия были начаты американским биохимиком X. Теминым, который еще в 1964 году обнаружил в клетках, зараженных РНК-содержащим вирусом саркомы Рауса, процесс синтеза ДНК, никогда не встречавшейся в незараженных этим вирусом клетках. Позднее X. М. Темин со своим сотрудником С. Мизутани и одновременно и независимо от них в другой лаборатории США Д. Балтимор открыли в составе частиц некоторых вирусов новый фермент – РНК-зависимую ДНК-полимеразу, уже само название которого говорит о его функции: с его помощью осуществляется построение молекул ДНК на матрице (основе) РНК. И именно такая вновь синтезированная ДНК интегрирует с ДНК клетки.
Эти открытия объясняли возможность интеграции геномов РНК-содержащих вирусов с геномом клетки и, таким образом, окончательно доказывали справедливость положений вирусо-генетической теории происхождения опухолей, тем более что фермент РНК-зависимая ДНК-полимераза (получивший название обратная транскриптаза или, еще короче, ревертаза) вскоре был обнаружен в составе частиц всех (!) РНК-содержащих опухолевых вирусов.
Итак, в основе превращения нормальной клетки в опухолевую лежит интеграция вирусных ДНК-копий, либо собственно вирусных, либо построенных на основе вирусной РНК, с клеточной ДНК. Поэтому, желая установить вирусную природу той или иной опухоли, в составе ДНК клеток ищут молекулы вирусных ДНК, для чего на сегодняшний день имеется несколько довольно чувствительных и точных методов. Следовательно, механизм интеграции ДНК вируса и клетки хозяина является правилом.
Но, как мы хорошо знаем, нет правил без исключений. Есть исключение и из правила превращения нормальной клетки в опухолевую. Речь идет о некоторых ДНК-содержащих вирусах, ДНК которых может персистировать (существовать длительное время) не будучи интегрированной с ДНК клетки, а в свободном (автономном) состоянии. Такие вирусные ДНК располагаются в клеточной цитоплазме, под их влиянием клетка превращается в раковую. В подобных клетках вирус продолжает существовать (персистировать) в виде изолированных вирусных ДНК, не связанных с клеточным геномом.
Однако возможностью интеграции геномов не исчерпываются механизмы вирусной персистенции. Так, несколько лет назад в одной из лабораторий изучали длительную персистенцию вируса ящура в организме теленка. При этом выяснился очень интересный факт: на протяжении года от этого теленка постоянно удавалось выделять вирус, который не размножался в культуре чувствительных клеток, если их после заражения культивировали при температуре 41 °С, т. е. соответствующей температуре тела теленка. Вместе с тем если такие же зараженные вирусом клетки инкубировали при температуре 37 °С, то вирус в них начинал активно размножаться и в конечном счете даже вызывал разрушение клеток.
Чем же вызвано подобное различие в поведении вируса в одних и тех же клетках? Оказалось, все дело в так называемых температурочувствительных мутантах. В результате мутационных изменений в геноме вируса иногда может быть затронут участок, который определяет синтез специального фермента – полимеразы, осуществляющего конструирование вирусных нуклеиновых кислот (геномов) при размножении вируса внутри клетки. При этом изменения в ферменте могут иметь весьма своеобразные последствия: фермент перестает работать или работает плохо при той температуре, при которой обычно вирус хорошо размножается. Но при этом, как оказалось, фермент успешно функционирует при более низкой температуре. Потому-то такие вирусные мутанты и назвали температурочувствительными.
Теперь представим себе, что группа клеток заражена температурочувствительным вирусом, но помещена в обычные для стандартного вируса температурные условия. Понятно, что такой мутантный вирус размножаться будет плохо, что приведет к накоплению в клетках вирусных «полупродуктов», образования же зрелого, полноценного вируса, имеющего в своем составе все необходимое, происходить не будет. Именно вирусные «полупродукты» будут персистировать в клетках. Нередко наличие в клетках таких «полупродуктов» может привести к совершенно неожиданным последствиям.
Вот один из примеров. Реовирус вызываету человека заболевание верхних дыхательных путей, диарею и жирный понос. Введение обычного штамма реовируса в мозг новорожденным крысятам вызывает у них скорое развитие острого энцефалита с высокой смертностью. Если же таких животных заражают температурочувствительным мутантом реовируса, то крысята в течение нескольких месяцев выглядят совершенно здоровыми, но затем у них медленно развивается гидроцефалия, которая постепенно прогрессирует и в конце концов заканчивается гибелью животных.
Наконец, говоря о механизмах вирусной персистенции в организме, нельзя забывать, что организм оснащен целым арсеналом достаточно разнообразных противовирусных средств, о которых уже шла речь выше. Одни из них всегда готовы к борьбе, всегда начеку – например, белые клетки крови (лейкоциты) и некоторые клетки мелких кровеносных сосудов, способные захватывать любые чужеродные для организма тела (в том числе и вирусы) и переваривать их. Другие «защитники» быстро образуются в зараженном организме в ответ на введение вируса, например противовирусные антитела и интерферон.
Отсюда мы вправе заключить, что формирование вирусной персистенции в организме представляет собой результат «неудач» в работе защитных механизмов организма хозяина. Случаи такой неудачи могут быть весьма разнообразными.
Действительно, некоторые вирусы, например, обладают выраженной способностью подавлять выработку антител, как это делает вирус гриппа или вирус лимфоцитарного хориоменингита. Другие, наоборот, способствуют накоплению порой колоссальных количеств антител, как, например, при заражении вирусом алеутской болезни норок, но… такие антитела не обладают способностью нейтрализовывать данный вирус. Третьи тормозят выработку интерферона или так изменяют его при этом, что такой интерферон не оказывает своего защитного действия. А, скажем, вирус инфекционной анемии лошадей после заражения животных поселяется и длительно персистирует только в макрофагах, и, само собой, им после этого уже не до того, чтобы очищать организм от вируса.






