2.1. Стрессорные кардиопатии
Стрессорная альтерация миокарда – причина гибели животных при иммобилизационном стрессе по Г. Селье. При моделировании иммобилизационного стресса одни авторы относят фатальный исход экспериментов за счет гиперпродукции стрессреализующих гормонов (катехоламины, кортикостероиды) [141], другие – стресслимитирующих (инсулин) [121].
В наших опытах отмечалась гибель 56 % крыс к 72 ч иммобилизации [27, 30], что не расходится с данными литературы. Практически в 100 % случаев наиболее вероятной причиной гибели животных при иммобилизационном стрессе является постепенно нарастающая стрессорная альтерация сердечной мышцы, приводящая к ее функциональной несостоятельности и развитию недостаточности кровообращения. Об этом свидетельствуют морфологические, а также биохимические признаки (нарушение окислительного фосфорилирования, активация ПОЛ, повышение текучести митохондриальных и микросомальных мембран кардиомиоцитов).
Тиамин, оптимизируя стресс-реакцию организма на действие неспецифических раздражителей, предотвращает гибель животных при иммобилизационном стрессе.
Из рис. I-1 видно, что у контрольных животных в стадии тревоги иммобилизационного стресса (1—12 ч) наблюдается резкий подъем содержания 11-ОКС в крови, который сохраняется на высоком уровне в течение 24 ч нервно-мышечного раздражения с последующим снижением в конце периода резистентности. В фазе истощения (48–72 ч) наблюдается новая волна стероидогенеза.
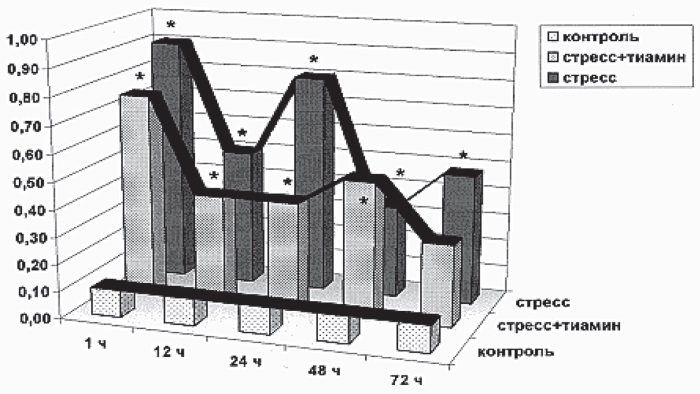
Рис. I-1. Стероидогенная реакция надпочечников в динамике хронического стресса до (черные столбики) и после (серые столбики) введения тиамина. Контроль – белые столбики. По оси абсцисс – срок наблюдения; по оси ординат – содержание 11-ОКС в крови, мкМ/л.
* Достоверные изменения – p < 0,05
Рис. I-2 демонстрирует динамику уровня ИРИ в крови тех же животных: существенный спад содержания гормона спустя 1 ч иммобилизации (стадия тревоги), затем стабилизация его на относительно низком уровне между 12–24 ч опыта с заметным уменьшением в конце стадии резистентности (24–48 ч) и резкий подъем к 72 ч (стадия истощения).
Длительность стадий иммобилизационного стресса определена в соответствии с данными [140]. Фазовая градация стресса традиционно приводится в терминах Г. Селье, хотя об истощении надпочечников при активации гормоносинтеза в корковом слое адреналовых желез при отсутствии тотальной деструкции кортикоцитов [16] в терминальную стадию раздражения говорить не приходится. Динамика 11-ОКС в течение 72 ч иммобилизационного стресса подтверждается результатами аналогичного эксперимента, проведенного ранее [6], а динамика ИРИ при хроническом истощающем раздражении – данными [121].
Если абстрагироваться от уровня нормы (интактные животные) и за точку отсчета взять стадию резистентности, то можно заметить, что начиная с 24 ч опыта содержание обоих гормонов в крови крыс изменяется однонаправленно. Это указывает на подчиненность инсулинового ритма кортикостероидному и находится в соответствии с известными данными о том, что гормоны коры надпочечников способны лимитировать инсулиногенез [13]. Реципрокное соотношение 11-ОКС и ИРИ в стадии тревоги (1—12 ч) свидетельствует о том, что инсулинотропное влияние кортикостероидов в первую и большую часть второй фазы иммобилизационного стресса, очевидно, нивелируется катехоламинами, которые подавляют секрецию инсулина, связываясь с ?-рецепторами ?-клеток поджелудочной железы [58]. Таким образом, не исключено, что при длительной иммобилизации животных динамика ИРИ в крови de facto определяется динамикой стресс-гормонов.
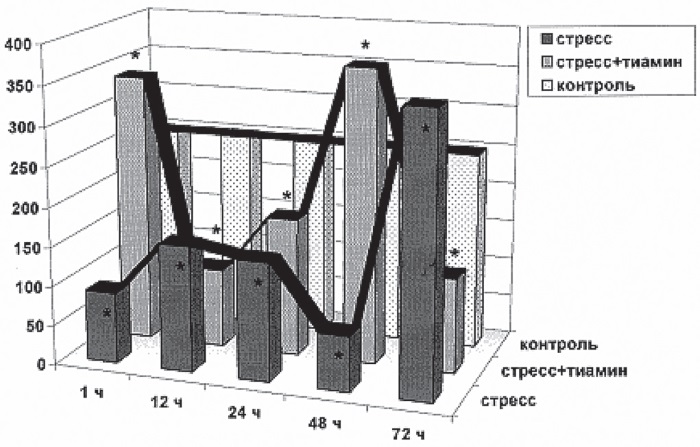
Рис. I-2. Содержание ИРИ (пкМ/л) в крови крыс в динамике хронического стресса до (черные столбики) и после (серые столбики) введения тиамина. Контроль – белые столбики. По оси абсцисс – срок наблюдения, по оси ординат – единицы измерения. * Достоверные изменения – p < 0,05
По мнению Л. Панина, при хроническом истощающем стрессе в фазу резистентности «продукция катехоламинов и глюкокортикоидов стремится к максимуму, а продукция инсулина к минимуму. Организм работает на пределе своих адаптационных возможностей и быстро переходит в стадию истощения, где происходит срыв регуляторных механизмов, в результате чего продукция инсулина может резко возрастать, развивается сильнейшая гипогликемия и организм погибает» [121].
Однако К. Судаков [153] считает, что в принятых условиях гибнут, прежде всего, стрессчувствительные животные от стрессорных кардиопатий – острой сердечной недостаточности или инфаркта миокарда. Так, в его опытах из 40 изученных беспородных крыс устойчивыми к иммобилизационному эмоциональному стрессу оказались 26, из них у 6 вообще не обнаружили изменений артериального давления, у 12 наблюдалось первичное его повышение с последующей стабилизацией. 14 крыс этой группы оказались предрасположенными к эмоциональному стрессу и погибли, проявляя различную динамику изменений артериального давления. У них при вскрытии были обнаружены массивные участки инфаркта миокарда [153].
Согласно Г. Селье, при длительной иммобилизации у животных нарушается Na+/K+ баланс в организме (развивается гипокалиемия), что предрасполагает к возникновению под влиянием продолжающейся стрессорной нагрузки неинфарктных некрозов сердечной мышцы, подобных тем, которые вызываются глюкокортикостероидами на фоне десенсибилизации с помощью ортофосфата натрия [141].
Следовательно, сегодня фактически существуют 3 гипотетических сценария гибели животных в терминальной фазе истощающего стресса: 1) срыв адаптации из-за функционального истощения надпочечников [140]; 2) фатальная гипогликемия, обусловленная нарушением (разбалансировкой) гуморальной регуляции, приводящей к гиперпродукции инсулина [121] и 3) несовместимые с жизнью стрессорные кардиопатии, обусловленные гиперактивностью симпато-адреналовой системы [153].
Первое допущение проверяли исследованием морфофункционального состояния кортикоцитов в течение всего периода иммобилизации животных, второе – измерением уровня глюкозы и ИРИ в крови крыс, а третье – электронномикроскопическим изучением альтерации кардиомиоцитов при непрерывном хроническом раздражении и применением тиамина, способного снижать продукцию кортикостероидов и катехоламинов при стрессе [17] и в силу этого являющегося потенциальным кардиопротектором. В последнем случае предполагалось, что если стрессорные кардиопатии на самом деле являются причиной гибели животных, то тиамин, как и любой антистрессор, должен увеличивать процент их выживания.
Результаты проверки показали, что на самом деле 3-суточная иммобилизация для значительной части животных (56 %) заканчивается фатально [16, 17, 27, 30]. При этом уровень 11-ОКС в строгом соответствии со сменой фаз ИС изменяется синусоидально: подъем – плато – снижение, т. е. до 48 ч опыта (начало фазы истощения) все происходит так, как предсказывает традиционная схема ОАС. А вот далее наблюдающийся подъем уровня 11-ОКС в предагональном состоянии (72 ч) ей явно противоречит.
Проведенные параллельно электронномикроскопические исследования ультраструктуры кортикоцитов позволяют понять, за счет чего это происходит. Действительно, как и предполагал Г. Селье, в фазу истощения в коре НП увеличивается количество изношенных секреторных клеток. На электронограммах видно, что в кортикоцитах, прилегающих к запустевшим, спавшимся или забитым продуктами микроклазматоза капиллярам, начинается краевая деструкция внутриклеточных структур, ответственных за различные этапы стероидогенеза: митохондрии разрушаются (рис. I-3), эндоплазматический ретикулум исчезает, цитозоль гомогенизируется (рис. I-4); хотя в дистальных отделах клетки органеллы еще сохраняют интактную ультраструктуру.
Наконец процесс дезинтеграции захватывает нуклеоплазму, ядерный хроматин фрагментируется, происходит кариорексис ядра (рис. I-5) и клетка перестает существовать как самостоятельная функциональная единица. Однако валовый рост деструкции клеточных элементов коры НП в терминальной стадии ИС (72 ч) не превышает 10 %, остальные 90 % кортикоцитов имеют совершенно нормальный вид.
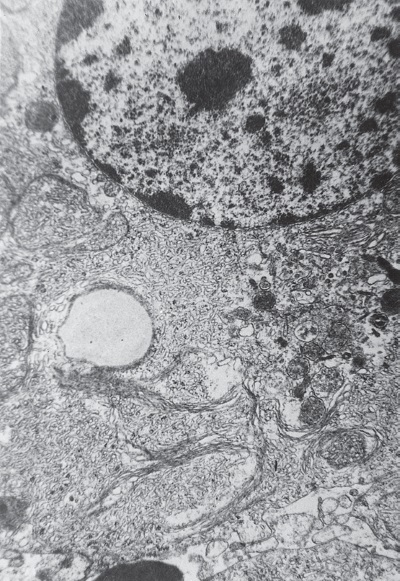
Рис. I-3. Деструкция митохондрий кортикоцитов при 72-часовом иммобилизационном стрессе. ? 71 000
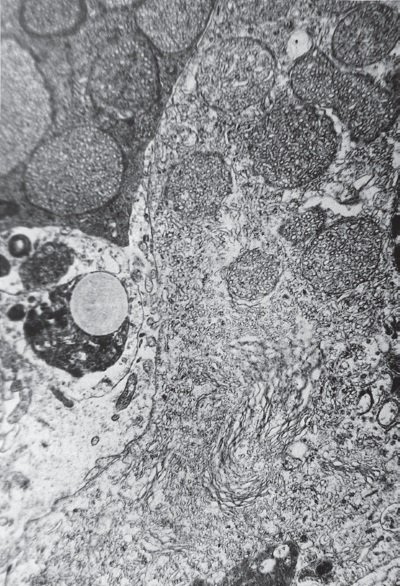
Рис. I-4. Дезорганизация цитозоля кортикоцитов при 72-часовом иммобилизационном стрессе. ? 71 000
Более того, к началу фазы истощения (48 ч) в клеточном спектре пучково-сетчатой зоны коры НП (рис. I-6) преобладают темные и очень темные кортикоциты с мощным регенераторным потенциалом, создающим громадный и легко мобилизуемый функциональный резерв стероидогенеза. К концу фазы истощения (72 ч) их количество несколько снижается за счет последовательной трансформации очень темных клеток в темные, затем в полутемные и наконец в секретирующие светлые клетки, которые и обеспечивают искомый прирост 11-ОКС в терминальной фазе стресса (рис. I-6).
Следовательно, ни о каком функциональном истощении НП здесь речь идти не может, так же как и том, что этот феномен может быть причиной гибели животных при ИС.
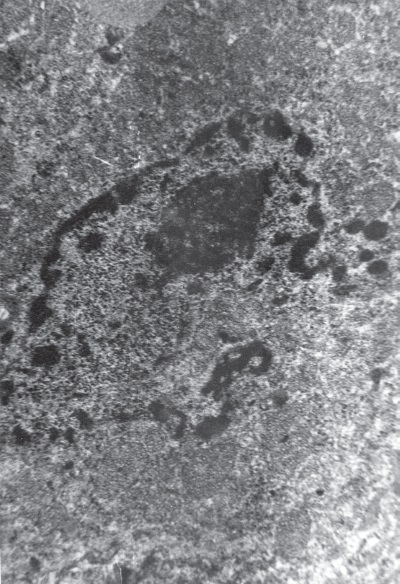
Рис. I-5. Кариорексис ядер кортикоцитов при 72-часовом иммобилизационном стрессе: выход гетерохроматина. ? 71 000 Рис.
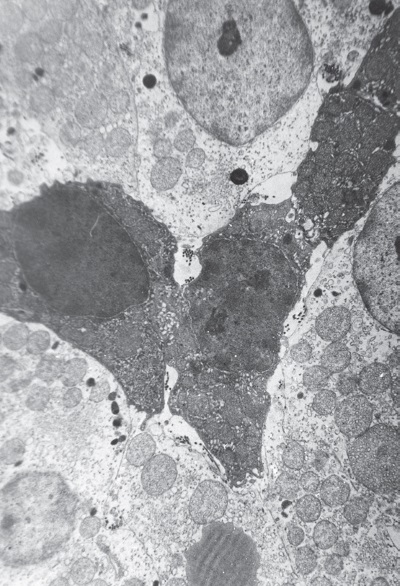
I-6. Клеточный спектр пучковой зоны коры надпочечника крысы при 72-часовом стрессе: очень темные и просветленные кортикоциты. ? 71 000
То же относится и к стресслимитирующей системе, роль которой в принятых условиях выполняет инсулин. В аварийную фазу ИС уже в первые часы уровень ИРИ в крови резко падает (рис. I-2) в результате прекращения инсулиногенеза при блокаде ?-рецепторов ?-клеток поджелудочной железы катехоламинами [58]. Развивается так называемый «транзиторный диабет напряжения», который в фазу резистентности (24 ч) плавно переходит в типичный стероидный диабет благодаря перераздражению инсулоцитов повышенным уровнем 11-ОКС, что не исключает развитие вторичной инсулярной недостаточности, т. е. истощение гормонобразовательной функции островкового аппарата к 48 ч опыта. Однако никакого истощения здесь тоже нет, поскольку в предагональном состоянии животных (72 ч) наблюдается достоверное повышение уровня ИРИ в крови. Такой всплеск инсулиногенеза Л. Панин считает результатом разбалансировки в системе гормональной регуляции гомеостаза, которая имеет фатальные последствия из-за развивающейся инсулиновой гипогликемии [121]. Измерение уровня глюкозы в крови с помощью ферментных электродов показало, что рост инсулинемии в терминальной стадии ИС (72 ч) сопровождается не гипо-, а гипергликемией. Несмотря на резкое повышение инсулинемии в фазу истощения (от 48 до 72 ч опыта – рост в 4 раза), содержание глюкозы в крови крыс в этот период достоверно не снижается (опыт 48 ч – 6,2 мМ/л (р>0,05); опыт 72 ч – 7,1 мМ/л (р>0,05); контроль – 8,1 мМ/л). Аналогичные данные были получены в эксперименте на крысах с применением летальной дозы ионизирующей радиации [123], где в предагональном состоянии (72 ч опыта) при всплеске инсулинопродукции (увеличение в 3–5 раз) содержание сахара в крови существенно возрастало (более чем в 1,5 раза) по сравнению с начальным периодом развития лучевой болезни (6 ч опыта) и в 1,3 раза по отношению к предыдущему сроку наблюдения (48 ч опыта). Следовательно, гибель половины животных к 72-му часу иммобилизации происходит не от гиперинсулинемии и сопутствующей гипогликемии, а от каких-то других причин.
Что касается гиперсекреции инсулина в терминальную фазу раздражения (рис. I-2), то скорее всего – это не разбалансировка в системе гуморальной регуляции гомеостаза [121], а признак начала функционирования инсулярного аппарата поджелудочной железы, освобождающейся к этому времени от катехоламинового блока. Восстановление же эндогенного (физиологического) механизма инсулиногенеза не может приводить к фатальной гипогликемии в принципе. Факт нормализации биологического ритма секреции инсулина в принятых условиях документируется тиаминовыми эффектами. В частности, на фоне введения тиамина уровень ИРИ в крови животных при иммобилизационном стрессе обнаруживает четкий 48-часовый ритм (рис. I-2), что соответствует данным литературы о 2-суточных осцилляциях содержания глюкозы в крови интактных крыс [117].
Экстраполируя эти результаты на динамику 11-ОКС, можно заметить, что кривая 1 (стресс) на рис. I-1 фактически демонстрирует развитие стероидогенной реакции на фоне «выключенного» инсулиногенеза (катехоламиновый блок ?-клеток поджелудочной железы), а кривая 2 (стресс + тиамин) – ту же реакцию, но на фоне активированного гормоносинтеза в инсулоцитах. Как видно, разница существенная и по амплитуде стероидогенной реакции (снижение), и по ее шагу (сдвиг влево).
Тиамин, активируя инсулинсинтетическую функцию поджелудочной железы [13], обеспечивает более раннее выхождение пика ИРИ в крови крыс при истощающем стрессе – сдвиг влево по временной шкале опыта с 72 ч на 48 ч иммобилизации (рис. I-2).
Синхронизация, т. е. удовлетворительное совпадение кривых 2 (стресс + тиамин) на рис. I-1 и I-2, свидетельствует о том, что при активированном инсулиногенезе стрессорный ритм 11-ОКС лимитируется уровнем ИРИ в крови крыс. Одновременно это означает, что антистрессорное действие тиамина, очевидно, опосредовано инсулином, который способен тормозить образование гормонов как в мозговом [355], так и корковом слое [177] надпочечников. Этим же объясняется и факт отсутствия гибели животных, получавших тиамин, при 3-суточной экспозиции иммобилизационного стресса, поскольку инсулин является мощным кардиопротектором [116]. Комбинацию инсулина с глюкозой давно используют в клинике для реабилитации больных с ишемическими повреждениями миокарда [82].
Таким образом, есть веские основания считать, что роль «киллеров» при истощающем стрессе выполняют стресс-гормоны (катехоламины и кортикостероиды), длительная гиперпродукция которых закономерно приводит к функциональной несостоятельности основной системы жизнеобеспечения – сердечно-сосудистой. Инсулину же здесь явно принадлежит хелперная функция поддержания жизнедеятельности. Данная констатация диктует стратегию выживания – применение антистрессорных средств, в том числе тиамина, оптимизирующего процессы гормоносинтеза в кортикальной и хромаффинной тканях надпочечников через механизм их инсулинового контроля.
Ультраструктура кардиомиоцитов. Поскольку при гистологическом исследовании сердец погибших животных, инфарктов или внеинфарктных некрозов миокарда не удалось обнаружить, основное внимание было уделено клеточному составу, морфологическим критериям нативности митохондриального и сократительного аппарата кардиомиоцитов, состоянию их саркоплазматического ретикулума, а также изменениям капиллярного русла во всех фазах развития стрессорной реакции, так как любые изменения функции сопровождаются морфологическими сдвигами [137].
В каждом случае оценивали ультраструктуру преобладающего типа клеток.
Контроль. Электронномикроскопически у интактных крыс в левом желудочке сердца выявляются кардиомиоциты двух типов: разносокращенные электронопрозрачные и равноплотные во всех частях светлые клетки (30 %) и релаксирующие полутемные клетки (70 %) с инвагинированным гиперхромным ядром, содержащим многочисленные впячивания нуклеолеммы и подстилающей широкой полосы маргинального гетерохроматина, слабоосмиофильными саркорплазмой, контрактильным аппаратом и очень темными митохондриями. Первые, которые находятся в явном меньшинстве, очевидно, осуществляют сократительную функцию миокарда в период относительного покоя, а вторые служат их функциональным резервом, легко мобилизуемым при рабочих нагрузках. По сравнению со светлыми клетками количество митохондрий в полутемных кардиомиоцитах заметно больше, а их площадь меньше. Мембраны всех органелл и их кристы практически не деструктированы, митохондриальный матрикс мелкозернист, одинаково темен и плотен. Все это свидетельствует о том, что энергообразовательный аппарат резервных клеток находится в спокойном состоянии и не участвует в сократительном акте (рис. I-7).
Митохондрии располагаются между миофибриллами в виде цепочек, иногда они образуют небольшие скопления. Большинство митохондрий имеют овальную, угловатую или вытянутую форму, двухконтурную наружную мембрану и значительное количество параллельно расположенных крист, пересекающих органеллы в поперечном направлении. Миофибриллы в полутемных клетках имеют типичное строение. На продольном срезе в них отчетливо дифференцируются диски А, I, полоса Н, мезофрагма М, иногда полосы N. Латентное состояние сократительного аппарата документируют отсутствие полос сокращения и одинаковая длина контрактильных элементов.
Границы саркомеров, ограниченные дисками Z, соседних миофибрилл совпадают друг с другом, сообщая кардиомиоцитам равномерную параллельную исчерченность. Канальцы Т-системы и гладкого саркоплазматического ретикулума без особенностей. Просвет капилляров расширен. Почти всегда в нем обнаруживаются эритроциты. Одни капилляры близко примыкают к кардиомиоцитам, другие отделены от них широким перикапиллярным пространством, которое плавно переходит в межклеточную щель. Эндотелий истончен, темен, содержит пиноцитозные пузырьки. Сморщенные гиперхромные ядра эндотелиальных клеток, отграниченные тонким ободком цитоплазмы и клеточной мембраной, выступают в просвет капилляра.
Фаза напряжения (1—12 ч опыта). В аварийную стадию иммобилизационного стресса в клеточном спектре левого желудочка преобладают светлые кардиомиоциты (80 %), резко снижается количество полутемных (18 %) и появляются единичные темные клетки (2 %).
Незначительная часть светлых кардиомиоцитов левого желудочка в эту фазу обнаруживает явную тенденцию к равномерному потемнению (переходные клетки), т. е. имеет признаки пересокращения, крайней формой которого является образование темных клеток. Большинство же светлых клеток находятся в состоянии умеренного сокращения или даже релаксации (рис. I-8). Все они имеют ядра вытянутой или округлой формы с уменьшенным количеством хроматина и просветленной нуклеоплазмой. Маргинация хроматина не выражена. Сохраняется двухконтурность ядерной мембраны. Канальцы Т-системы и саркоплазматического ретикулума несколько расширены.
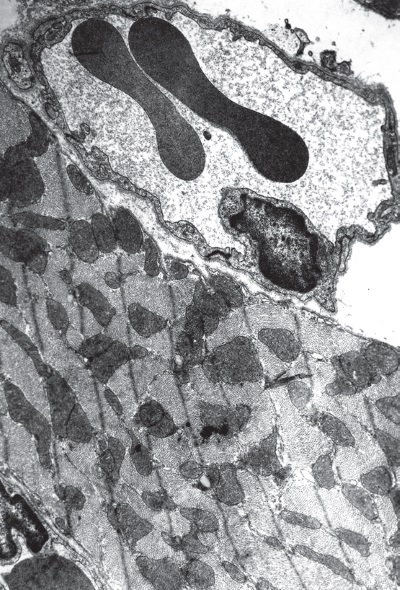
Рис. I-7. Миокард левого желудочка интактной крысы. Полутемные кардиомиоциты. ? 71 000

Рис. I-8. Миокард левого желудочка крысы при иммобилизационном стрессе. Фаза напряжения (1 ч опыта). Светлые кардиомиоциты. ? 71 000
Миофибриллы слегка отечны, местами волокнисты. Диски Z хорошо видны. Вставочные диски утолщены, имеют расширенные щели, границы их несколько расплывчаты. Между миофибриллами расположены двухконтурные митохондрии, имеющие овальную или вытянутую форму. Наблюдается повсеместное набухание органелл и самих клеток. Электронная плотность митохондрий, саркоплазмы, нуклеоплазмы и миофибрилл кардиомиоцитов выравнивается. В светлых клетках по сравнению со всеми остальными количество митохондрий найменьшее, площадь их – найбольшая, кристы сильно деструктированы, матрикс просветлен, пятнисто вымыт или вакуолизирован. Все эти особенности позволяют оценить наблюдаемую картину как отражающую состояние гиперфункции митохондриального аппарата.
Просвет большинства капилляров сужен как за счет набухания собственного эндотелия, так и сдавливания их тесно прилегающими гипертрофированными кардиомиоцитами. Перикапиллярные пространства и межклеточные щели как самостоятельные объемные образования между светлыми клетками не выявляются.
Фаза резистентности (24 ч опыта). В эту стадию развития стрессорной реакции кардиомиоциты в левом желудочке примерно поровну (по 40 %) представлены двумя типами клеток: переходными и светлыми. Причем первые от вторых отличаются меньшим отеком саркоплазмы и снижением степени набухания митохондрий, т. е. уплотнением их матрикса (рис. I-9, I-10). Количество митохондрий значительно превышает их число не только в светлых, но и в полутемных клетках. Встречаются очень большие скопления митохондрий. Большинство органелл имеют округлую форму и сохраняют наружную мембрану, двухконтурность которой в некоторых участках теряется. Матрикс митохондрий плотный, мелкогранулярный, в местах отсутствия крист гомогенизирован. Гомогенизация матрикса в различных органеллах колеблется от незначительных участков до всей митохондрии. Кристы несколько извилисты, но, как правило, они сохраняют параллельность и пересекают органеллы, соединяясь с наружной мембраной противоположных сторон. Почти во всех митохондриях часть крист разрушена и эти участки гомогенизированы.
В переходных клетках активируется процесс репродукции митохондрий. Он осуществляется путем деления и почкования органелл. О том, что митохондрии делятся, не сливаются, свидетельствует факт точного пространственного совпадения противолежащих крист соседних органелл. При почковании возникают перетяжки, истончение которых приводит к слиянию наружных мембран, а последующий их разрыв – к появлению дочерних митохондрий. Миофибриллы находятся в состоянии умеренного сокращения без очагов дезорганизации миофиламентов. В миофибриллах нередко увеличен диск I. Извитость вставочных дисков заметно увеличивается. Ядра кардиомиоцитов округлые, содержат повышенное количество хроматина, который концентрируется под нуклеонемой.
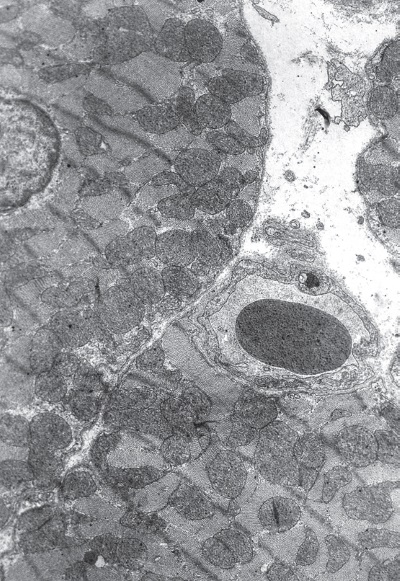
Рис. I-9. Миокард левого желудочка крысы при иммобилизационном стрессе. Фаза резистентности (24 ч опыта). Слабо сокращенные переходные кардиомиоциты. ? 71 000 Рис.
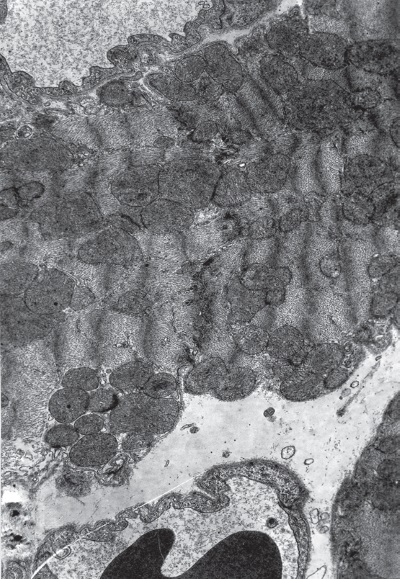
I-10. То же, что и на рис. I-9. Фаза резистентности (24 ч опыта). Сильно сокращенные переходные кардиомиоциты. ? 71 000
Капилляры отделены от сарколеммы кардиомиоцитов широкими прекапиллярными пространствами, переходящими в межклеточные щели различной ширины. Просветы большинства капилляров резко расширены и, как правило, заполнены свободно циркулирующими эритроцитами. Эндотелий капилляров уплощен, клетки – умеренной электронной плотности. Пиноцитоз выражен незначительно. Ядра овальной формы, нуклео-плазма просветлена в центре.
Судя по всему, переходные клетки – это интенсивно работающие кардиомиоциты, которые вместе со светлыми обеспечивают сократительную функцию сердца в фазу резистентности иммобилизационного стресса. Одновременно они накапливают регенераторный потенциал (деление митохондрий) и обнаруживают признаки перехода в другое функциональное состояние (обезвоживание и равномерное увеличение электронной плотности цитоструктур), т. е. трансформации в темные клетки (20 %). На рис. I-9, I-10 отчетливо видна динамика этого процесса. Степень обезвоживания клеток можно оценить по изменению сарколеммы, которая вначале имеет ровный пузыревидный контур, отграничивающий разбухшие кардиомиоциты, а в конце процесса по мере сокращения отечности саркоплазмы и выпячивания в межклеточные щели конгломератов митохондрий сарколемма принимает вид аркад, дублирующих контуры прилегающих органелл.
Конец фазы резистентности – начало фазы истощения (48 ч опыта). Морфологически (по клеточному составу и состоянию сосудистого русла) начало и конец фазы истощения четко различаются. К 48 ч опыта в клеточном спектре левого желудочка сердца явно преобладают темные кардиомиоциты, а к 72 ч – светлые, с множественными явлениями деструкции митохондриального и сократительного аппарата.
Темные клетки (рис. I-11), по сути, это пересокращенные переходные, которые характеризуются максимальной осмиофилией всех субклеточных образований. Сарколемма в темных клетках образует крутые аркады, в которых располагаются митохондрии, а саркоплазма практически не выявляется. В тех участках, где митохондрии прилежат к сарколемме, последняя теряет двухконтурность. Повышенная электронная плотность темных миокардиальных клеток зависит как от структуры органелл, так и от топографии последних.
Темные кардиомиоциты содержат компактные гиперхромные ядра с преобладанием конденсированного хроматина, часто имеющие изрезанные контуры. Количество митохондрий в них значительно больше, чем в светлых клетках, и они располагаются не параллельными с миофибриллами рядами, а в виде скоплений в различных участках клетки. Между миофибриллами можно видеть крупные митохондриальные агломераты, занимающие нередко все поле зрения. Причем органеллы здесь настолько тесно расположены, что другие структуры клетки почти незаметны (рис. I-11). Большое количество митохондрий располагается также в околоядерной зоне, под сарколеммой вблизи капилляров, у межклеточных щелей. Большинство митохондрий находится в конденсированном состоянии, матрикс их плотный, нередко частично гомогенизирован. Разные органеллы содержат различное количество крист. Многие митохондрии увеличены в размерах, но это увеличение не имеет характера набухания: матрикс таких органелл не просветлен, межкристные пространства не расширены, а кристы не только не деформированы и не разрушены, а напротив, их становится больше и они как бы более плотно упакованы в теле митохондрии. Во многих миофибриллах наблюдаются полосы пересокращения и различной величины участки гомогенизации миофиламентов. В субсарколемной зоне иногда возникают небольшие участки расплавления миофибрилл.
Увеличение количества темных клеток в миокарде при 48-часовой нагрузке растяжением животных можно рассматривать как одну из компенсаторных реакций сердца в ответ на повреждающий фактор. Появление в этих условиях темных кардиомиоцитов с резко увеличенным числом митохондрий подтверждает известную точку зрения на темные клетки как источники материальных и энергетических ресурсов, которые возникают в связи с напряженной попеременной деятельностью сократительных элементов и активацией в них процессов внутриклеточной регенерации [137].
Другой важной особенностью, характеризущей данный период стресса, является повышение гетерогенности клеточного пула левого желудочка.
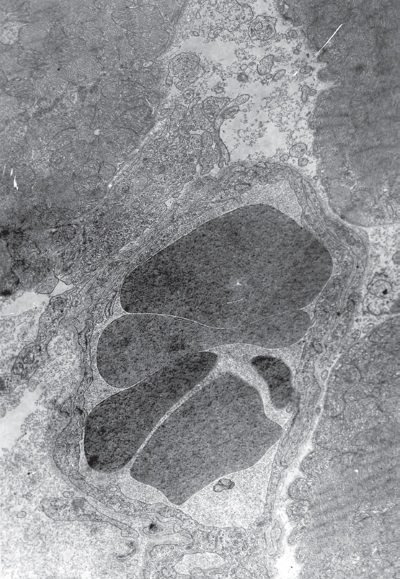
Рис. I-11. Миокард левого желудочка крысы при иммобилизационном стрессе. Конец фазы резистентности – начало фазы истощения (48 ч опыта). Темные кардиомиоциты. ? 71 000
Если в фазу напряжения иммобилизационного стресса практически не выявлялись переходные и темные клетки, в фазу резистентности – полутемные, то к началу фазы истощения клеточный спектр кардиомиоцитов представлен всеми типами клеток (темные – 50 %, светлые – 30 %, переходные – 10 %, полутемные – 10 %). Усиленная трансформация одних клеток в другие свидетельствует о повышении скорости оборачиваемости клеточного цикла, что, очевидно, также имеет в принятых условиях приспособительное значение. Процесс «просветления» темных кардиомиоцитов сопровождается постепенным нарастанием количества свободной саркоплазмы и разобщением структурных компонентов. В ядрах происходит превращение гетерохроматина в диффузный хроматин. В саркомерах появляются изотропные диски, а протофибриллы располагаются более рыхло. Становится заметной саркотубулярная система. Так возникают полутемные клетки, которые по мере продвижения по клеточному циклу трансформируются в светлые с ортодоксальными митохондриями и просветленным матриксом. Указанные структурные сдвиги свидетельствуют о вступлении «покоящихся» темных и полутемных кардиомиоцитов в фазу повышенной функциональной активности.
Сократительную функцию миокарда в конце фазы резистентности и начале стадии истощения иммобилизационного стресса (48 ч опыта) обеспечивают главным образом светлые и отчасти переходные клетки. В данный период по сравнению с фазой напряжения (1—12 ч опыта) количество светлых клеток в левом желудочке существенно снижено. Соответственно падающая на них рабочая нагрузка пропорционально увеличивается, что приводит к повышенному износу контрактильного и митохондриального аппарата светлых кардиомиоцитов. Увеличению степени деструкции сократительных элементов левого желудочка способствует ухудшение их кислородного обеспечения в результате спазма интрамуральных артериол, вызванного сокращением гладкомышечного слоя сосудистой стенки (рис. I-12). Волна нарушения микроциркуляции захватывает и капиллярное русло.
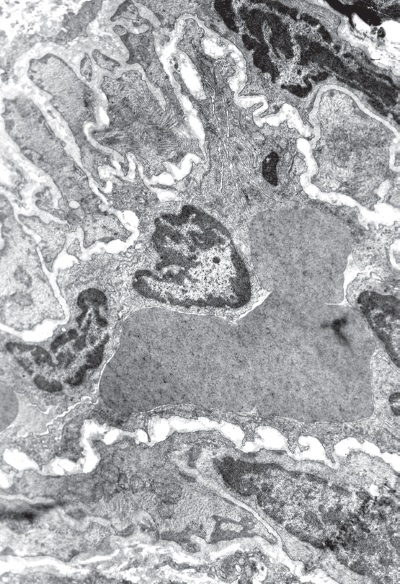
Рис. I-12. То же, что и на рис. I-11. Конец фазы резистентности – начало фазы истощения (48 ч опыта). Спазм интрамуральных артериол. ? 71 000
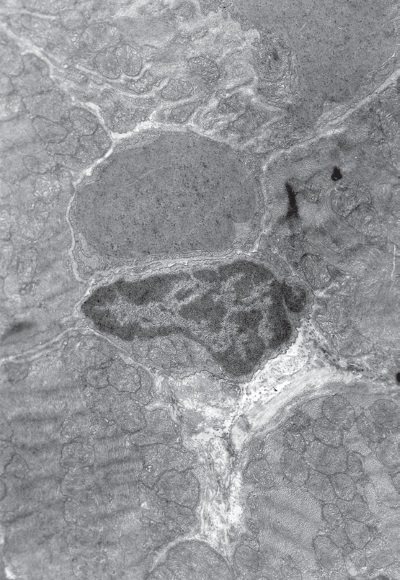
Рис. I-13. То же, что и на рис. I-11. Конец фазы резистентности – начало фазы истощения (48 ч опыта). Сдавливание капилляров пересократившимися темными клетками. ? 71 000
Повсеместно наблюдаются явления гемостаза, обусловленные сдавливанием капилляров пересократившимися темными клетками (рис. I-13) и гипертрофированными перицитами, которые в сложной связи своих отростков охватывают эндотелиальную трубку в виде своеобразной муфты (рис. I-14). Свой вклад в сужение просвета капилляров в данный срок опыта вносят и сами эндотелиальные клетки, способные к периодическому набуханию под влиянием нервных импульсов. В схеме двигательной иннервации кровеносных капилляров важная роль отводится перицитам, где находятся окончания симпатических нервов, которые воспринимают, трансформируют и далее передают нервный импульс через свои отростки на эндотелиальную клетку по типу электрического синапса [168]. Если принять гипотезу электрического синапса, то из рис. І-15 видно, что последний может возникать и без посредничества перицитов, поскольку нервные терминали непосредственно граничат с эндотелиальными клетками.
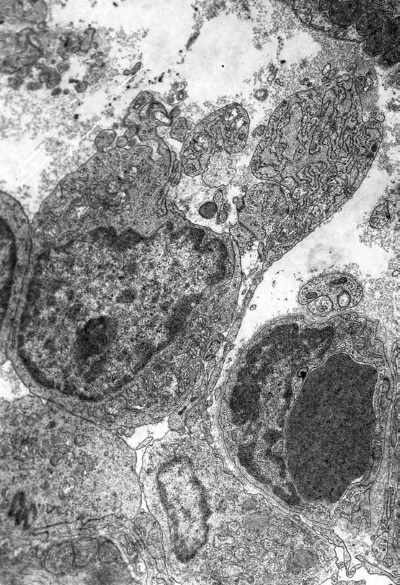
Рис. I-14. То же, что и на рис. I-11. Конец фазы резистентности – начало фазы истощения (48 ч опыта). Сдавливание капилляров гипертрофированными перицитами. ? 71 000
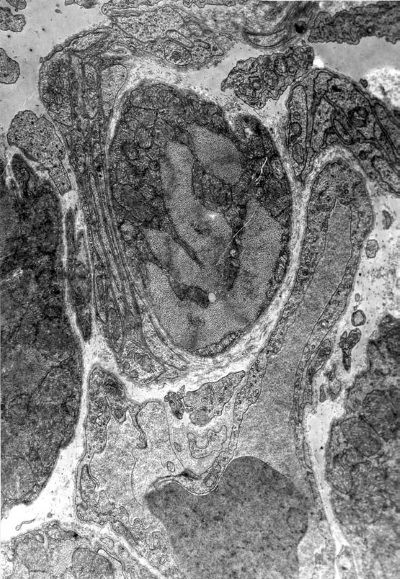
Рис. I-15. То же, что и на рис. I-11. Конец фазы резистентности – начало фазы истощения (48 ч опыта). Окончания нервных терминалей. ? 71 000
Согласно гипотезе, нервный импульс, направленный в сторону эндотелиальной клетки, вызывает деполяризацию ее плазмалеммы, что может способствовать потере или накоплению клеткой жидкости, которая, по всей вероятности, проникает через микропоры в плазмалемме [168]. По мнению автора гипотезы, на электронномикроскопических снимках и при прижизненных наблюдениях можно обнаружить набухшие эндотелиальные клетки, которые полностью закрывают просвет капилляра. Вслед за набуханием эндотелиальной клетки через несколько секунд можно видеть ее спадение. Такая периодичность, ведущая в первом случае к сужению просвета кровеносного капилляра и освобождению просвета для движения крови во втором случае, физиологически оправдана и не может быть осуществлена без участия нервной системы.
Фаза истощения (72 ч опыта). В терминальной стадии иммобилизационного стресса типовая гетерогенность кардиомиоцитов практически исчезает. Как и в самом начале развития стрессорной реакции, в левом желудочке в этот период доминируют светлые клетки (90 %; 10 % составляют полутемные клетки), что, очевидно, является результатом длительной перегрузки ультраструктурных элементов сердца. Однако в отличие от фазы напряжения (1—12 ч опыта), где светлые клетки были в основном однородными и различались только по степени набухания, здесь отмечается их выраженная внутрипуловая гетерогенность, обусловленная различной выраженностью деструкции митохондриального и контрактильного аппарата. В сарколемме таких кардиомиоцитов появляются множественные дефекты. На отдельных участках, особенно в тех, к которым близко прилежат капилляры, сарколемма становится размытой. В большинстве мышечных клеток отмечается отек саркоплазмы, особенно в субсарколемной зоне. Наряду с набухшими, но сохранившими нативную ультраструктуру кардиомиоцитами (30 %) – светлые клетки 1-го типа (рис. I-16) – появляются светлые клетки 2-го типа, в которых почти все митохондрии разрушены (20 %). Миофибриллы в них в основном фрагментированы на уровне дисков I, но имеются и большие участки гомогенизации, разволокнения и разрыва миофиламентов. Многие диски Z смещаются по отношению друг к другу в лежащих рядом миофибриллах (рис. I-17). В светлых клетках 3-го типа (40 %) большинство митохондрий в состоянии либо выраженного, либо умеренного набухания, матрикс их очагово просветлен. Резко уменьшено количество крист, имеющиеся кристы фрагментированы, хаотично расположены, в центре митохондрий они превращаются в гомогенную слабоосмиофильную массу. Наружные мембраны митохондрий теряют двухконтурность либо на всем протяжении, либо на значительных участках. Немало митохондрий полностью гомогенизированных, а также резко набухших, лишенных матрикса и крист.
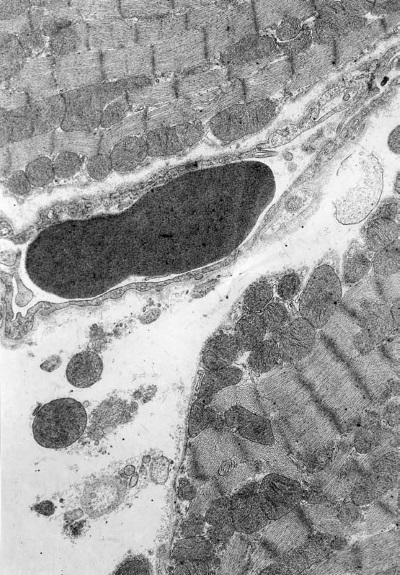
Рис. I-16. Миокард левого желудочка крысы при иммобилизационном стрессе. Фаза истощения (72 ч опыта). Деструктивные кардиомиоциты первого типа. ? 71 000
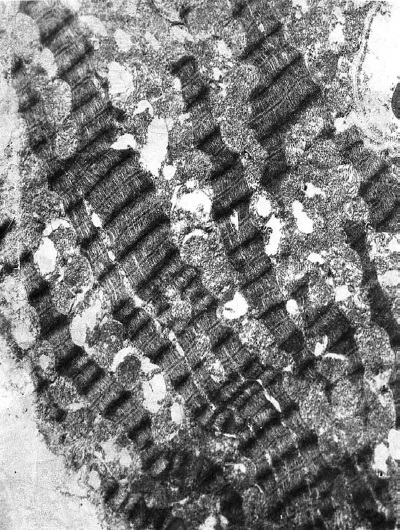
Рис. I-17. То же, что и на рис. I-16. Фаза истощения (72 ч опыта). Деструктивные кардиомиоциты второго типа. ? 71 000
Примечательна мозаичность изменения митохондрий: в одной и той же клетке имеются митохондрии с различной степенью изменений – от умеренного набухания и неравномерного расположения крист до полной деструкции органелл (рис. I-18). В миофибриллах отмечается появление множественных участков деструкции. На продольных срезах отчетливо видно, что вначале происходит расплавление тонких миофиламентов. Затем возникают очаги их деструкции на протяжении нескольких саркомеров. Разделительные диски Z исчезают. В этих участках сохраняются лишь обрывки толстых миофиламентов и остатки дисков I (рис. I-18). В других участках происходит гомогенизация миофибрилл. В очагах гомогенизации заметны обрывки миофиламентов, которые затем подвергаются расплавлению. Наблюдается значительная вариабельность в степени повреждения между различными мышечными клетками этого типа. В некоторых кардиомиоцитах в областях разрушения миофибрилл видна гомогенная масса миофиламентов, в то время как другие области имели нормальный или почти нормальный вид. В очагах тяжелого повреждения клетки спектр изменения был довольно широким – от конденсации до полного лизиса миофибрилл.
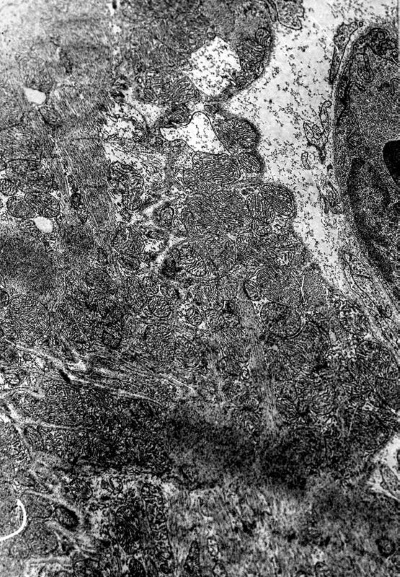
Рис. I-18. То же, что и на рис. I-16. Фаза истощения (72 ч опыта). Деструктивные кардиомиоциты третьего типа. ? 71 000
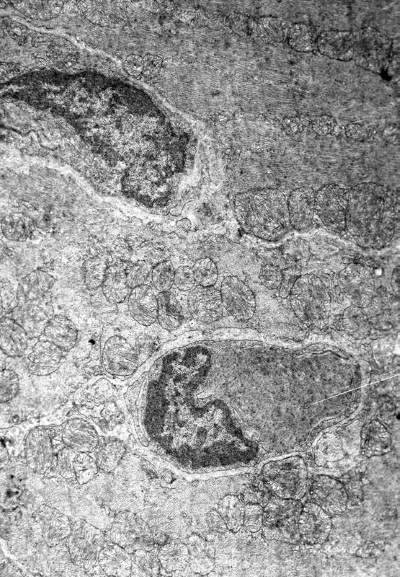
Рис. I-19. То же, что и на рис. I-16. Фаза истощения (72 ч опыта). Лизис кардиомиоцитов. ? 71 000
На периферии таких поврежденных участков миофибриллы часто пересокращены и подвержены процессу гомогенизации и лизиса (рис. I-19). Сопряжение кардиомиоцитов нарушается за счет расхождения плазматических мембран, образующих вставочные диски и их фрагментации (рис. I-19), что характерно для декомпенсации сердца, сопровождающейся развитием фибрилляции желудочков [122]. Показателем функциональной перегрузки светлых кардиомиоцитов 3-го типа является резкое расширение канальцев саркоплазматического ретикулума вплоть до образования крупных вакуолей и цистерн (рис. I-18). Повреждения саркотубулярной системы, играющей важную роль в распространении возбуждения по миокардиальной клетке, могут привести к замедлению распространения импульса и возникновению блоков проведения, т. е. к прогрессированию возникшей в миокарде фибрилляции [122]. Кроме того, перерастяжение и частичное разрушение канальцев саркотубулярной системы может стать основой ослабления сократительной силы сердца, так как приходящее в миофибриллы возбуждение не будет реализовываться их сокращением. Такой механизм может лежать в основе развития сердечной недостаточности. Существуют три основные причины, обусловливающие сердечную недостаточность: 1) нарушение функции саркоплазматического ретикулума поглощать, накапливать и отдавать Са2+, участвующий в акте сокращения, 2) дефицит энергии и 3) функциональная несостоятельность контрактильного аппарата кардиомиоцитов [164]. Морфологически сочетание действия этих негативных факторов в принятых условиях документируется резким расширением канальцев саркоплазматического ретикулума, деструкцией митохондрий и миофибрилл (рис. I-17, I-18). По сути дела тот же сценарий развития стрессорных кардиопатий, предложенный Ф. Меерсоном, заложен в механизме «кальциевой триады»: накопление Са2+ в кардиомиоцитах из-за неполадок в системе внутриклеточного транспорта катиона может привести к недостаточности сердца в результате возникновения кальциевых контрактур, активации фосфолипаз и особенно протеаз, разрушающих диски миофибрилл, и нарушения окислительного фосфорилирования в нагруженных Са2+ митохондриях [98].
Действительно, как видно из рис. I-18, в светлых кардиомиоцитах 3-го типа налицо типичные признаки кальциевого повреждения ультраструктуры: имеются контрактурные полосы пересокращения миофибрилл с гомогенизацией содержимого, лизис саркомеров, начинающийся с Z дисков, накопление в межкристном пространстве сильно набухших митохондрий осмиофильных включений ортофосфата кальция. Считается, что перегрузка миокардиальных клеток кальцием является главной причиной альтеративных сдвигов не только при классических кальциевых (адреналиновых, изопротереноловых) некрозах, но и при гиперфункции, гипертрофии, ишемии (неглубокой), гипоксии, стрессорных повреждениях, большинстве некоронарогенных болезней сердца [246].
Повреждения сарколеммы и изменения саркоплазматического ретикулума могут приводить к сбоям в работе кальциевой помпы и Na-Ca-ионообменного механизма [97], т. е. к прогрессирующему накоплению содержания катиона в кардиомиоцитах в результате проникновения избыточного Са2+ внутрь клетки из межклеточного пространства через образовавшиеся дефекты в плазмалемме [246], блокирования процесса откачки кальция из клетки и слабости механизмов его внутриклеточной утилизации. В итоге в клетках образуется массивная кальциевая перегрузка [239], которая морфологически проявляется накоплением в матриксе митохондрий электронноплотных гранулированных осадков фосфата кальция [239], появлением участков пересокращения саркомеров и расширением канальцев саркоплазматического ретикулума [167]. С указанными изменениями связывают затруднение в проведении нервных импульсов, нарушения ионного транспорта, развитие незавершенной диастолы [97] и снижение сократимости миокарда при хроническом дефиците насосной функции сердца [311]. Независимо от конкретной причины накопления избыточного Са2+ в саркоплазме этот процесс неизбежно приводит к одному и тому же эффекту – несостоятельности энергообеспечения кардиомиоцитов за счет снижения генерации энергии в митохондриях, гиперактивации Са-зависимых АТ-Фаз и истощению АТФ в местах его использования [218], что составляет патогенетическую основу развития сердечной недостаточности.
Ядра светлых кардиомиоцитов всех 3 типов – округлые, без признаков маргинации хроматина, которую обычно расценивают как показатель ухудшения дренажной функции саркоплазмы [109]. В межклеточных и перикапиллярных пространствах возле светлых клеток всегда много однооболочечных митохондрий с гомогенизированным матриксом, разнокалиберных первичных и вторичных лизосом, которые служат морфологическим маркером стресс-реакции [251].
В завершающей стадии иммобилизационного стресса (72 ч опыта) в миокарде левого желудочка встречаются три типа капилляров. Просветы капилляров первого типа широкие, содержат эритроциты (рис. I-16). Цитоплазматические отростки эндотелиальных клеток гофрированы, пиноцитоз умеренный. Просветы капилляров второго типа, тесно прилегающих к разрушающимся кардиомиоцитам, почти полностью закрыты набухшими эндотелиальными клетками или эритроцитарными тромбами (результат гемостаза), в их ядрах количество хроматина увеличено (рис. I-19). Наконец, третий тип капилляров отличается тем, что эндотелиальные клетки здесь образуют длинные цитоплазматические клапаны, куда часто смещаются гиперхромные ядра. Эти отростки достигают противоположной стенки капилляра и делят его просвет на несколько полостей. Вокруг таких капилляров очень много поврежденных митохондрий, различного типа лизосом, эритроцитов (рис. I-20). Базальный слой капилляров первого и третьего типа не изменен, а второго – сужен.
По сравнению со стадией относительного покоя (контрольные животные), где в клеточном спектре левого желудочка сердца преобладают полутемные кардиомиоциты, содержанием каждой стадии иммобилизационного стресса является некий главный (определяющий энергетику) процесс, который лимитирует появление преобладающего типа клеток: в фазу напряжения – набухание (светлые); в фазу резистентности – деление (переходные) и регенерация (темные); в фазу истощения – деструкция (деструктивные клетки). Трансформация одного типа клеток в другой в рамках замкнутого жизненного цикла кардиомиоцитов: светлые – переходные – темные – полутемные – светлые является проявлением механизма стабилизации энергетики при прогрессировании гиперфункции сердца в условиях непрекращающегося раздражения. Деструктивные светлые клетки – это необратимо поврежденные кардиомиоциты, сошедшие с орбиты жизненного цикла.
Набухание митохондрий в первой стадии стресса (1– 12 ч опыта) является наиболее быстрой реакцией этих органелл на те требования, которые предъявляются к клетке новыми условиями функционирования. Быстрая трата энергии приводит к изменению соотношения компонентов адениловой системы внутри митохондрий, что, в свою очередь, обусловливает изменение ионного баланса по обе стороны митохондриальных мембран и степень гидратации митохондрий. Поступление в них воды вызывает увеличение объема митохондрий, расправление крист и общее увеличение энергообразующей поверхности. Выработка энергии усиливается и происходит восстановление нарушенного энергетического гомеостаза клетки [160]. Следовательно, набухание митохондрий можно расценивать как показатель их гиперфункции. Если оно превышает критический уровень, выход энергии резко снижается в результате перерастяжения внутренних митохондриальных мембран и пространственного разобщения энергообразующих комплексов. Чрезмерное набухание митохондрий в фазе истощения иммобилизационного стресса (72 ч опыта) следует расценивать как переход адаптивно-приспособительной реакции, мобилизующей для контрактильных элементов миокарда дополнительные энергетические резервы, в патогенетическую, приводящую к необратимым нарушениям структурной целостности органелл и их гибели (рис. І-17).
Степень набухания митохондрий регулируется прежде всего уровнем оксигенации клетки. У стрессированных животных площадь капиллярного русла миокарда заметно снижается в начальную (1 ч – фаза тревоги) и терминальную (72 ч – фаза истощения) стадию иммобилизационного стресса. В эти же сроки на начальном и завершающем пиках содержания стресс-гормонов в крови (катехоламинов [121] и 11-ОКС [16]) в кардиомиоцитах наблюдается набухание митохондрий, особенно выраженное в фазу истощения, что указывает на аварийную гиперфункцию органелл в условиях дефицита кислорода, когда осуществляется энергообеспечение только специализированной функции сократительных клеток.
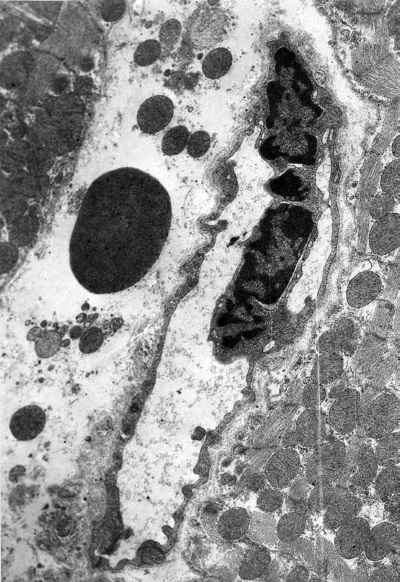
Рис. I-20. То же, что и на рис. I-16. Фаза истощения (72 ч опыта). Перегородки (клапаны) в просвете капилляра. ? 71 000
Деление и регенерация митохондрий во второй стадии стресса возникают тогда, когда нагрузка, падающая на миокард, не устранена и нарастающее действие повреждающего фактора приводит к усилению морфологических изменений в миокарде. Вначале на первый план выступают явления, связанные прежде всего с восстановлением количества митохондрий в кардиомиоцитах (24 ч опыта), а затем основными становятся процессы регенерации митохондрий, утративших кристный материал (48 ч опыта). Однако при непрерывном раздражении животных это восстановление никогда не бывает полным. Даже при увеличении общего количества митохондрий и содержания в них крист в каждой отдельной митохондрии количество крист не достигает исходного уровня, что является причиной постоянной гиперфункции органелл и их ускоренного разрушения [122].
Нарастающий энергетический дефицит вызывает необходимость включения в каждый цикл функционирования кардиомиоцита все большего количества митохондрий и тем самым прогрессивно сокращается возможность их полноценной регенерации. Таким образом, несмотря на усиление процессов регенерации митохондрий, во второй стадии одновременно увеличивается и процесс изнашиваемости органелл. Эффективность генерации энергии в митохондриях отдельных кардиомиоцитов постепенно снижается, и для поддержания насосной функции сердца в каждом цикле его сокращения включается все большее количество миокардиальных клеток, в которых
при этом начинают накапливаться деструктивные изменения. Дефектное энергообеспечение увеличивает дисбаланс между деструктивными и биосинтетическими процессами, что, в свою очередь, приводит к постепенному исчезновению компенсаторно-приспособительных реакций и нарастанию изменений, обусловливающих в последующем развитие сердечной недостаточности.
Деструкция митохондрий и миофибрилл кардиомиоцитов в третьей заключительной стадии стресса – это срыв приспособительных механизмов, а прогрессивно нарастающее нарушение биоэнергетических процессов в сердце, приводящее к появлению деструктивных светлых клеток, можно рассматривать как коллапс энергетики миокарда. Энергетическое истощение лежит в основе развития и так называемого комплекса изнашивания гипертрофированного сердца [96], поскольку хроническая недообеспеченность миокардиальных элементов приводит их к гибели. На этой основе закономерно возникает сердечная недостаточность, а явления укорочения эффективного рефрактерного периода миокарда создают предпосылки для возникновения различных нарушений ритма сердца – от экстрасистолии до смертельной фибрилляции желудочков [96].
Таким образом, судя по морфологическим сдвигам ультраструктуры кардиомиоцитов, непосредственной причиной гибели животных в терминальной фазе истощающего иммобилизационного стресса, по всей вероятности, является сердечная недостаточность, развивающаяся на фоне фатальной несостоятельности энергообеспечения сердечной мышцы.
Биоэнергетика сердца. Отражением стрессорной реакции на метаболическом уровне может быть прежде всего адаптационная перестройка энергетического обмена, а именно изменение функционирования систем генерации и потребления энергии [121]. Общее представление о состоянии энергообразовательной функции митохондрий сердца при стрессе можно получить уже исходя из данных сравнительного исследования дыхания органелл с помощью стандартной полярографической техники.
Обработка полярограмм включала определение скорости дыхания митохондрий после последовательных добавок 10 мМ сукцината (V0), 100 мкМ АДФ (V3 и V4), 400 мкМ динитрофенола (V5). Рассчитывались также величины дыхательного контроля ДКл и ДКч по Ларди – Вельману (V3/V2) и по Чансу – Уильямсу (V3/V4).
Полученные результаты приведены в табл. I-1. Истощающий иммобилизационный стресс по Г. Селье [140] отчетливо повышает скорость дыхания митохондрий сердца во всех метаболических состояниях. Причем увеличение окислительной активности митохондрий сопровождается ухудшением энергетической регуляции дыхания. Динамические параметры дыхательной цепи при стрессе, например коэффициент усиления, отражающий эффективность сопряжения дыхания и фосфорилирования (ДКл), и дыхательный контроль в отрегулированном состоянии, отражающий степень восстановления энергизации митохондрий после рабочей нагрузки (ДКч), существенно снижены по сравнению с нормой, что характеризует работу органелл в режиме утомления с прогрессирующим переходом к низкоэнергетическому состоянию
[70]. Явными проявлениями низкоэнергетического сдвига в принятых условиях являются: ослабление энергетической регуляции дыхания (рост дыхания в состоянии покоя (V0) и снижение ДКл), рост дыхания в состоянии V2 и признаки повреждения митохондрий: относительное снижение окисления сукцината в активном состоянии (V3), в фазе истощения иммобилизационного стресса (72 ч), которое in vitro не устраняется глютаматом, и снижение ДКч больше, чем ДКл, во все сроки опыта.
Иммобилизационный стресс на фоне тиамина не сопровождается признаками повреждения митохондрий сердца: скорость окисления сукцината в терминальной фазе стресса (72 ч) продолжает нарастать без резкого ухудшения энергетической регуляции дыхания (имеет место относительный рост ДКл, а также одинаковое увеличение ДКл и ДКч), которое наблюдается у стрессировавшихся крыс. Скорее всего, в принятых условиях тиамин действует как антистрессор: витаминзависимое снижение амплитуды стероидогенной реакции (рис. I-1) автоматически ограничивает степень активации сукцинатдегидрогеназы стрессорными гормонами [39]. Поскольку добавка ЩУК-устраняющего субстрата (глютамата) in vitro повышает скорость окисления ЯК митохондриями, выделенными из сердца животных, получавших тиамин во все фазы стресса, здесь возможно и другое объяснение.
Таблица I-1.
Влияние тиамина (Т) на окислительную и фосфорилирующую функцию сердца крыс в динамике ИС
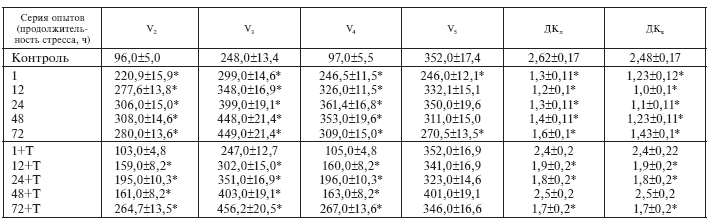
* Достоверные изменения – p < 0,05.
Согласно Г. Селье, существует 2 типа адаптационных механизмов – кататоксические, ответственные за активное сопротивление раздражителю, и синтоксические, обеспечивающие пассивную устойчивость и сосуществование с патогенным воздействием [312]. Примером перехода от кататоксических реакций к синтоксическим является тиаминзависимое ограничение окисления сукцината митохондриями сердца крыс (V3) в динамике истощающего иммобилизационного стресса (табл. I-1). Фактически это ограничение представляет собой синтоксическую реакцию, которая и обеспечивает сосуществование с раздражителем, повышая пассивную устойчивость за счет снижения активных реакций.
Для иммобилизационного стресса показано, что на уровне митохондрий ЩУК-обусловленное ограничение дыхания выполняет функцию синтоксической реакции, предупреждая кататоксическую гиперактивацию окисления сукцината [70]. Известно, что увеличение доли ЯК в общем окислении обеспечивает повышенные энергетические запросы при активности. Однако эта компенсаторная реакция подобно адаптационным реакциям на уровне организма может становиться чрезмерной и повреждающей. В условиях повышенного содержания жирных кислот и ионов Са2+, характерных для стресса, повышается проницаемость мембран митохондрий и возрастает интенсивность окисления субстратов. При этом активируется сукцинатдегидрогеназа и гиперактивное окисление сукцината становится источником дальнейшего повреждения мембраны [69]. На энергизованных митохондриях печени сукцинат обычно оказывает стабилизирующее действие [15], а повреждение органелл при его окислении наблюдается на интенсивно метаболизирующих низкоэнергизованных объектах, таких, как митохондрии сердца голубя и митохондрии патологического сердца человека [148]. Считается, что повреждающее действие ЯК, проявляющееся снижением или потерей дыхательного контроля, обусловлено накоплением протонов и гиперактивным транспортом ионов кальция в митохондрии.
Кальцификация тканей характерна для стресса, и она базируется на гиперактивном окислении сукцината. Интенсификация окисления этого субстрата и транспорта Са2+ в митохондрии наблюдается в ткани сердца человека при тяжелых формах сердечной недостаточности [148]. Залповый импорт кальция в кардиомиоциты после ишемизации сердца ответствен за повреждение миокарда [98]. В таких условиях блокирование дыхательной цепи цианидом предотвращает чрезмерное поступление внешнего кальция в кардиомиоциты и их повреждение [69]. Поскольку тотальное ингибирование дыхательной цепи не может быть использовано в целостном организме для in vivo профилактики повреждения митохондрий сердца крыс, при длительной иммобилизации животных некоторые авторы применяли введение антагониста катехоламинов – серотонина, который снижает дыхание тканевых препаратов [81] предположительно за счет индукции ЩУК-механизма ограничения окисления сукцината и тем самым обеспечивает защиту органелл от стресса [69]. Это допущение согласуется с повышением уровня серотонина в организме при таких типичных проявлениях хронического стресса, как язвенная болезнь желудка и 12-перстной кишки, раздражении электротоком, возбуждении, хирургических операциях и других стрессобусловленных патологических состояниях [81]. Не исключено, что инициация ЩУК-ограничения окисления сукцината митохондриями сердца при стрессе является характерным моментом действия любого антистрессора, в том числе и тиамина.
В присутствии динитрофенола, вызывающего полное разобщение дыхания и фосфорилирования, потребление кислорода митохондриями животных в терминальной фазе истощающего стресса (72 ч опыта) снижено в 1,75 раза (табл. I-1). Это означает, что изменения, выявляемые в митохондриях, изолированных из сердца животных при длительной иммобилизации, выражаются не только в нарушении сопряжения окисления с фосфорилированием, но и в нарушении самого окисления, т. е. транспорта электронов в дыхательной цепи. В соответствии с современными представлениями такие нарушения могут быть обусловлены повреждением липидного бислоя митохондриальных мембран продуктами перекисного окисления липидов (ПОЛ), а также чрезмерной активацией фосфолипаз сердечной мышцы и детергентного действия избытка жирных кислот, возникающего при стрессе в результате повышения секреции катехоламинов [98]. Как видно из табл. І-2, с увеличением экспозиции иммобилизации крыс содержание в сердце продуктов ПОЛ (диеновые конъюгаты, малоновый диальдегид) неуклонно возрастает (с максимумом к 72 ч опыта), а уровень антиоксидантной защиты кардиомиоцитов (активность каталазы, супероксиддисмутазы и содержание эндогенного токоферола) пропорционально падает, т. е. имеет место прогрессирующее нарушение исходной сбалансированности между ферментными системами генерирования и детоксикации липопероксидов за счет стрессзависимого снижения антиоксидантного статуса организма [138].
Предварительное введение нетоксичных доз природных и синтетических антиоксидантов (токоферол, ионол) предупреждает типичную для тяжелого ЭБС активацию ПОЛ в мышце сердца, мозге и других органах. В результате ингибирования ПОЛ не развивается повреждений мембран кардиомиоцитов, нарушения работы Санасоса, окислительного фосфорилирования в митохондриях, избыточной потери миокардом ферментов, транзиторного повреждения и последующей репарации ДНК; предотвращаются депрессия сократительной функции миокарда и постстрессорное снижение устойчивости сердца к гипоксии. Эти факты свидетельствуют о том, что активация ПОЛ действительно составляет ключевое звено патогенетической цепи стрессорной альтерации кардиомиоцитов. Блокирование ПОЛ, устраняющее возможность накопления гидроперекисей липидов в мембранных структурах кардиомиоцитов, очевидно, приводит к стабилизации их липидного бислоя и этот мембранотропный эффект составляет суть кардиопротекторного действия антиоксидантов [98]. В механизме снижения уровня ПОЛ под влиянием антиоксидантов, содержащих гидроксильные группы фенольного типа, наряду с осуществлением их скевенджерной функции «ловушек» супероксидных радикалов важную роль может играть и собственная антистрессорная активность препаратов.
Предварительное введение ионола (2-6-дитретбутил-4-метилфенола) заметно уменьшает амплитуду стрессорной реакции в ответ на эмоционально-болевое воздействие: убыль катехоламинов в надпочечниках у стрессированных животных уменьшается в 2 раза, а подъем концентрации кортикостерона в плазме, обычно наблюдаемый после ЭБС, практически отсутствует [98].
Отсюда не исключено, что в спектре защитного действия любого стресслимитирующего фактора должны присутствовать мембранопротекторные эффекты, реализуемые через снижение уровня ПОЛ, пропорциональное степени индуцированной резистентности организма к данному виду стресса.
Действительно, как видно из табл. І-2, тиамин, который не содержит гидроксильных групп фенольного типа и практически не обладает антиоксидантными свойствами [149], но способный эффективно снижать уровень гормоносинтеза в секреторных клетках коры надпочечников [13] и хромаффинных клетках мозгового вещества адреналовых желез [17] в условиях развития реакции напряжения, тем не менее существенно ограничивает степень активации ПОЛ в миокарде при иммобилизации крыс. Истощающий стресс на фоне тиамина приводит к заметно меньшему приросту продуктов ПОЛ (ДК, МДА) в сердце и соответственно меньшему снижению антиоксидантного потенциала кардиомиоцитов (активности каталазы, супероксиддисмутазы и содержания токоферола), чем у животных, не получавших витамин В1. Поскольку аналогичное действие на антиоксидантный статус организма в принятых условиях оказывает адреналэктомия (табл. I-2), есть все основания считать, что ограничение ПОЛ под влиянием тиамина лимитируется соответствующим снижением уровня стрессорных гормонов в крови крыс, т. е. по сути является антистрессорным эффектом.
Таблица I-2.
Влияние тиамина (Т) на про– и антиоксидантный статус кардиомиоцитов в динамике ИС
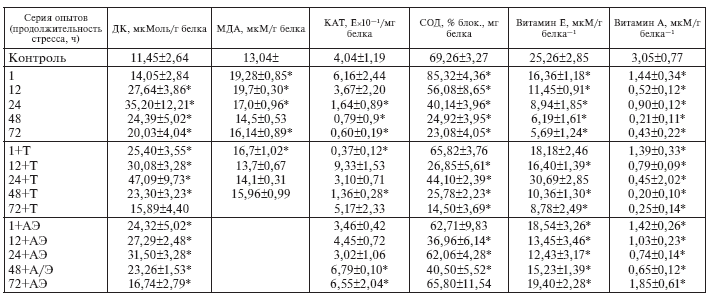
* Достоверные изменения – p < 0,05.
Существуют различные варианты гипотезы перекисной гибели клеток. В модели Ф. Меерсона, адаптированной к кардиомиоцитам, важная роль отводится «липидной триаде», элементы которой при стрессе формируют своеобразный порочный круг с взаимным усилением друг друга: активация ПОЛ – лабилизация лизосом кардиомиоцитов – освобождение лизосомальных фосфолипаз – гидролиз встроенных в мембрану фосфолипидов – образование свободных жирных кислот (ЖК) и лизофосфатидов – нарушение упорядоченности бислойных мембран – увеличение проницаемости их мембран для Са2+ – активация фосфолипаз, что в конечном итоге приводит к необратимому повреждению сарколеммы и внутриклеточных мембранных структур и апоптозу [98].
Известно, что эффективность функционирования биологических мембран существенно зависит от физического состояния их липидов. Одним из информативных методов оценки физического состояния липидов биологических мембран является метод флуоресценции, с использованием флуоресцентных зондов [52]. Мы изучали физические свойства (микровязкость) свободных (липидный бислой) и связанных с белками (анулярных) липидов мембран митохондрий и эндоплазматического ретикулума кардиомиоцитов сердца крыс при иммобилизационном стрессе до и после введения тиамина.
Использование гидрофобного флуоресцентного зонда пирена, инкорпорированного в зону жирнокислотных цепей фосфолипидов, позволяет оценить физические свойства мембраны в местах локализации зонда.
Причем перекрывание спектров поглощения зонда и эмиссии триптофанилов дает возможность селективно, за счет индуктивно-избирательного переноса энергии (ИРПЭ), возбуждать молекулы пирена, расположенные в непосредственной близости к мембранным белкам (анулярные липиды). Возбуждение непосредственно самой молекулы пирена позволяет характеризовать состояние бислойных или свободных липидов [114].
Как видно из табл. I-3, микровязкость свободных липидов (липидного бислоя) микросомальных мембран кардиомиоцитов при хроническом стрессе уменьшается. Минимальное значение регистрируется уже после первого часа иммобилизации и остается на таком уровне до 24 ч опыта. В области анулярных липидов микровязкость также уменьшается, но минимум достигается после 12-часового стрессирования. Микровязкость бислойных липидов митохондриальных мембран изменяется аналогичным образом.
Уменьшение микровязкости окружения зонда, встроенного в липиды микросомальных и митохондриальных мембран, свидетельствует об увеличении их текучести. Повышение текучести мембран мозга и сердца [72] наблюдается при старении и гипокинезии. Одной из причин этого явления может быть изменение состава фосфолипидов. Показано, что причиной увеличения текучести липидного бислоя мембран саркоплазматического ретикулума кардиомиоцитов является увеличение количества длинноцепочечных полиненасыщенных жирных кислот в составе фосфолипидов [68].
Таким образом, иммобилизационный стресс приводит к изменению физических свойств липидного матрикса мембран эндоплазматического ретикулума и митохондрий сократительных клеток миокарда. Текучесть липидного бислоя и анулярных липидов увеличивается в течение 72-часового стрессирования и более выражена для митохондриальных мембран (табл. I-3). Вероятно, эти изменения могут привести к нарушению функциональных свойств мембран, в частности транспорта ионов кальция [75].
Приведенные в табл. І-3 результаты свидетельствуют, что у животных, не получавших тиамин, микровязкость свободных липидов микросомальных мембран в миокарде после одного часа стрессирования уменьшается в 4 раза, а у витаминизированных – только в 1,5 раза. В дальнейшем на протяжении всего эксперимента она остается практически на одном уровне.
Таблица І-3.
Влияние тиамина (Т) на физические свойства липидов митохондриальных мембран кардиомиоцитов крыс при ИС
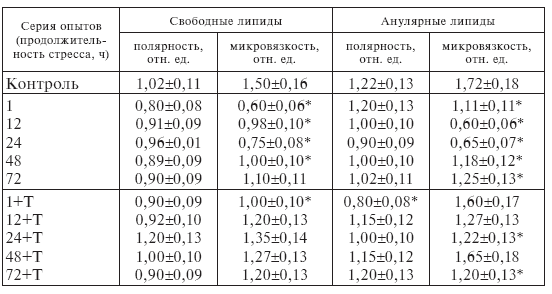
* Достоверные изменения – р < 0,05.
Аналогичные изменения микровязкости наблюдаются для анулярных липидов микросом, свободных и анулярных липидов митохондрий. Во все сроки иммобилизации тиамин снижает текучесть липидов микросомальных и митохондриальных мембран (для анулярных липидов максимальное снижение наблюдается через 1 ч и 48 ч опыта). Очевидно, его введение приводит к стабилизации флуктуаций жирных кислот в составе фосфолипидов изучаемых мембран за счет снижения их фосфолипазного расщепления [84].
Фосфолипидам митохондрий обычно отводят роль материала, скрепляющего глобулы белков, которые образуют структурную основу митохондриальной мембраны. Разрушение комплекса белок – фосфолипид – белок будет иметь следствием снижение той жесткости мембраны, которая необходима для поддержания высокого гидростатического давления в матриксе и препятствует осмотическому набуханию органелл [350]. Предполагается, что молекулы фосфолипидов-«скрепок» составляют ту небольшую часть фосфолипидного фонда митохондриальных мембран, которая специфически подвержена
действию фосфолипазы митохондрий [202]. Гидролиз одной из сложноэфирных связей между глицерофосфатом и жирной кислотой, катализируемой этой фосфолипазой, приводит к распаду тройного комплекса, расхождению исходно фиксированных белковых глобул, уменьшению жесткости мембраны в целом и набуханию митохондрий [350].
Таким образом, повышение текучести мембран митохондрий кардиомиоцитов при длительной иммобилизации крыс (табл. І-3) свидетельствует о набухании органелл, а тиамин, уменьшая текучесть, препятствует набуханию во все сроки опыта за счет повышения резистентности организма животных к стрессу и соответственно снижения его мембранотропного влияния.
В последней связи важно было выяснить, как тиаминзависимая стабилизация митохондриальных мембран отражается на функциональной активности органелл. Для этого оценивали скорость потребления кислорода суспензией митохондрий в разобщенном ДНФ (V5) и активном (V3) состояниях при использовании в качестве субстрата окисления сукцината. Оказалось, что при одночасовой и двухсуточной экспозиции иммобилизационного стресса (сроки, где проявлялось максимальное мембраностабилизирующее действие тиамина – табл. І-3) по абсолютным значениям показателей V5 и V3 митохондрии сердца крыс, получавших тиамин, и митохондрии контрольных животных не отличались друг от друга (табл. I-1). Не исключено, что в принятых условиях гарантом стабилизации мембран основных цитоплазматических структур сократительных клеток миокарда является антистрессорное действие тиамина, которое обусловливает снижение актуальности раздражения для кардиомиоцитов, т. е. уменьшает «падающую» на них рабочую нагрузку при иммобилизационном стрессе.
Медиатором высокоамплитудного набухания митохондрий кардиомиоцитов при иммобилизационном стрессе может быть Ca2+, который через активацию митохондриальных фосфолипаз [98] способен лабилизировать мембраны органелл, увеличивая их проницаемость для молекул воды. Оценивая полученные результаты (табл. І-3) с позиций этого допущения, следует иметь в виду, что действие стресса на сердце является по существу адренергическим и через систему адренорецепторов приводит к увеличенному вхождению Ca2+ в кардиомиоциты [138]. Последнее играет важную роль в положительном инотропном эффекте катехоламинов и при умеренном стрессе оказывается транзиторным, так как благодаря нормальному функционированию мембранных механизмов ионного транспорта избыток Ca2+ быстро удаляется из саркоплазмы. При истощающем стрессе, сопровождающемся повреждением мембран и катионных насосов, удаление Ca2+ из саркоплазмы может оказаться нарушенным. Чрезмерное накопление Ca2+ в кардиомиоцитах имеет два следствия. Во-первых, он может активировать совокупность процессов, составляющих липидную триаду [98], и таким образом замыкает порочный круг, углубляющий повреждение миокарда. Во-вторых, избыток Ca2 обладает собственным повреждающим действием, которое приводит к разобщению окисления и фосфорилирования в митохондриях, активации митохондриальных фосфолипаз и миофибриллярных протеаз, угнетению процесса расслабления миофибрилл вплоть до развития необратимых контрактур и некробиоза [100]. Нарушение поглощения, депонирования и выброса кальция кардиомиоцитами считается важнейшей причиной развития сердечной недостаточности [164].
Из материалов опытов следует, что процесс приспособления сердца к иммобилизационному стрессу протекает в жестких временных рамках развития общего адаптационного синдрома: фаза напряжения (1—12 ч), фаза резистентности (12–48 ч), фаза истощения (48–72 ч) [15]. Синфазность стероидогенной (рис. I-1) и кардиогенной (табл. І-1, І-2, І-3) составляющих реакции напряжения указывает на их взаимообусловленность. Смысловая адекватность фазовой динамики стресса и гормональноиндуцированной кардиопатии, а также возможность логической экстраполяции характеристик одного процесса на другой подчеркивают фундаментальную патогенетическую общность обоих состояний.
Стабилизирующие эффекты тиамина в отношении окислительного фосфорилирования в митохондриях кардиомиоцитов (табл. І-1) и текучести их мембран (табл. І-3) наблюдаются на пике содержания стрессорных гормонов в крови животных (рис. І-1), т. е. в фазе напряжения (1 ч) и фазе истощения (48 ч) иммобилизационного стресса. Это подтверждает стрессобусловленность отмеченных сдвигов и антистрессорный характер действия витамина В1. В принятых условиях тиамин понижает потолок реагирования организма на чрезвычайные раздражители, переводя основные системы жизнеобеспечения (в том числе гипофизадреналовую и сердечно-сосудистую) с гиперергического на более низкий уровень перекрестной адаптации, отвечающий степени раздражителя. Патофизиологически это проявляется оптимизацией морфофункциональных параметров мембранных структур кардиомиоцитов и увеличением выживаемости животных при длительном иммобилизационном стрессе.






